Dettagli
- Definizione
- Patologia
- Clinica
- Caratteristiche radiologiche
- Immagine 01
- Immagine 02
- Immagine 03
02.37 – [Radiodiagnostica] Fibrosi polmonare
Definizione
- La patologia fibrotica polmonare si caratterizza per la formazione di un eccesso di tessuto fibrotico nel polmone e rientra a far parte del grande capitolo delle malattie polmonari interstiziali diffuse.
- Tale condizione patologica è caratterizzata da un ispessimento dell’interstizio peribroncovasale, perilobulare e subpleurico (grosso interstizio) e/o dell’interstizio parenchimale inter-alveolo-capillare (piccolo interstizio), a causa della sostituzione fibrotica della matrice connettivale.
- La fibrosi polmonare può essere localizzata, segmentaria, lobare o interessare l’intero polmone mono o bilateralmente.
Patologia
- Tantissime sono le patologie in cui si ha deposito di tessuto fibroso a livello dell’interstizio polmonare, così come i quadri anatomopatologici che ne sono alla base: molte rappresentano differenti entità patologiche in quanto caratterizzate da specifiche alterazioni anatomopatologiche in aggiunta alla fibrosi e tali malattie sono:
- malattie granulomatose: TBC, micosi, sarcoidosi ed alveoliti allergiche estrinseche, che si caratterizzano per la formazione di granulomi a livello interstiziale con caratteristiche di nodularità;
- collagenopatie e malattie autoimmuni: sclerodermia, artrite reumatoide, polimiosite, sindrome di Sjogren, in cui l’interessamento polmonare è solitamente collaterale in un quadro sistemico di malattia;
- malattie congenite: fibrosi cistica;
- pneumoconiosi: silicosi, asbestosi, ecc;
- ipertensione post-capillare prolungata ed edema polmonare cronico, in cui si verifica un’organizzazione del liquido edematoso negli spazi interstiziali perché il drenaggio linfatico non è in grado di smaltire il liquido che ristagna a livello polmonare;
- fibrosi post-attinica dovuto all’esposizione a radiazioni come avviene in radioterapia;
- linfangite carcinomatosa per diffusione neoplastica dall’interstizio polmonare a livello linfatico;
- fibrosi da farmaci: metotrexate, bleomicina, busulfano;
- polmoniti interstiziali idiopatiche: polmonite interstiziale usuale (UIP), polmonite interstiziale acuta (AIP), polmonite interstiziale non specifica (NSIP), polmonite organizzata criptogenetica (COP), polmonite interstiziale desquamativa (DIP), bronchiolite respiratoria con polmonite interstiziale associata (RBILD), polmonite interstiziale linfocitaria(LIP);
- insulto acuto severo: ARDS, infezione polmonare, danno alveolare diffuso;
- condizioni croniche: polmonite cronica da ipersensibilità, polmonite eosinofila cronica;
- malattie con fibrosi negli stadi terminali: emosiderosi polmonare idiopatica, sindrome di Goodpasture, proteinosi alveolare, ARDS, istiocitosi X.
Clinica
- Sebbene possano essere molte le cause che determinano la fibrosi polmonare la sintomatologia che si associa a questa condizione è sempre più o meno la stessa, ovvero un quadro del tutto aspecifico caratterizzato:
- da tosse secca;
- dispnea ingravescente (prima a riposo poi sotto sforzo man mano che aree sempre più ampie di parenchima vengono interessate);
- astenia;
- calo ponderale;
- insufficienza respiratoria di tipo restrittivo.
- Il diffuso impegno interstiziale, oltre ad agire sugli scambi gassosi, riducendone l’efficienza, porta anche ad alterazioni della normale vascolarizzazione polmonare con associata ipertensione polmonare secondaria ad aumento delle resistenze vascolari ed un quadro terminale di cuore polmonare cronico.
- Essendo una malattia che generalmente impiega molti anni a diventare clinicamente manifesta, al momento della diagnosi il paziente presenta solitamente già un quadro restrittivo ampiamente sviluppato con clinica e test di funzionalità respiratoria (spirometria ed emogasanalisi) in grado di confermare la diagnosi di fibrosi polmonare. Tali test sono generalmente sufficienti (molto sensibili) ma poco specifici per la diagnosi eziologica del processo e per la capacità di localizzazione topografica dell’interstiziopatia.
Caratteristiche radiologiche
- Compito della diagnostica per immagini è identificarne l’aspetto prevalente della fibrosi, in modo da definire il quadro anatomopatologico integrando i dati clinici e i il dato bioptico, di quantificare l’estensione del coinvolgimento interstiziale e seguire il quadro nella sua evoluzione a lungo termine.
RX
- I reperti mostrati possono anche essere del tutto normale: nelle due proiezioni AP e LL forniscono una visione globale del parenchima ed interstizio polmonari, potendo evidenziare eventuali alterazioni dell’interstizio.
- Generalmente l’interstizio polmonare non è evidenziabile all’Rx del torace se non nella sua componente vascolare quindi, l’eventuale presenza di lesioni interstiziali sarà subito riconoscibile, grazie anche ad alcuni aspetti peculiari:
- ispessimento della trama polmonare interstiziale localizzata o diffusa a sede peri-bronco-vascolare e/o centrale e/o periferica;
- distribuzione di tipo reticolare o lineare;
- molteplicità delle alterazioni, in quanto solitamente sono malattie che hanno carattere sistemico o polmonare in toto e quindi difficilmente si localizzano ad un’area circoscritta;
- persistenza di aree a normale diafania polmonare, dal momento che non vengono generalmente coinvolti gli spazi alveolari, almeno nelle forme iniziali ed intermedie.
- Data la lenta insorgenza e sviluppo della malattia, nei casi in cui non sia nota la sua eziopatogenesi, il quadro radiologico che si ha il più delle volte è del tutto aspecifico, con gli elementi appena riportati, e non consente una diagnosi eziologica su base radiografica.
- Ci sono poi diversi tipi di lesione interstiziale che dovranno essere identificate:
- opacità lineari: dovute ad un interessamento dell’interstizio perilobulare che va incontro ad ispessimento, con localizzazione preferenziale alle basi polmonari e in sede sub-pleurica e prendono il nome di strie di Kerley;
- opacità reticolari: l’impegno interstiziale è maggiore rispetto alle opacità lineari e sono dovute a malattie che si sviluppano nell’interstizio peribroncovasale e lobulare, spesso in rapporto al coinvolgimento delle strutture linfatiche (come avviene nella linfangite carcinomatosa). Le pareti dei grossi bronchi ed i tessuti peribronchiali si ispessiscono creando immagini a binario o cuffie peribronchiali (bronchial cuffing). Abbiamo una maglia di linee di diverso spessore (da meno di 1 mm a pochi mm) distribuite irregolarmente nei campi polmonari e che si uniscono intrecciandosi;
- opacità nodulari: sostenute da malattie che si sviluppano nell’interstizio peribroncovasale e in quello lobulare sotto forma micro e/o macronodulare (malattie granulomatose come la sarcoidosi o la TBC) o che giungono al polmone tramite l’albero arterioso (metastasi, tubercolosi miliare) o l’albero bronchiale (silicosi). Si tratta di noduli multipli di forma rotondeggiante, a margini netti, delle dimensioni di 1-10 mm.
- Oltre a queste caratteristiche, tipiche di tutte le interstiziopatie e che generalmente diventano conclamate solo nelle fasi avanzate della malattia, ci sono poi dei quadri peculiari che caratterizzano ogni singola patologia, soprattutto se la loro scoperta avviene nelle fasi precoci, quando ancora hanno un pattern di impegno interstiziale peculiare. Questi elementi specifici si presentano in alcune malattie interstiziali configurando alcune entità definite.
TC
- Tutti i reperti radiografici descritti in precedenza possono essere riconoscibili in fase precoce tramite la TC del torace ad alta risoluzione (HRTC), quando la funzionalità respiratoria può non essere ancora significativamente alterata.
- Inoltre la TC del torace ad alta risoluzione permette anche lo studio dell’interstizio intra-lobulare ed una migliore definizione del danno anatomico e pertanto tale metodica riveste un ruolo importante per:
- diagnosi precoce: nei pazienti in cui non vi è evidenza radiografica di lesioni interstiziali, ma è presente un quadro clinico compatibile con interstiziopatia, la TC del torace ad alta risoluzione potrà mettere in evidenza la presenza di lesioni in fase precoce;
- valutazione quantitativa e qualitativa del danno, in modo da valutare l’estensione della malattia e la sua reale gravità (enfisema paracicatriziale, bronchiectasie, atelettasie, ecc);
- valutazione di patologie concomitanti (linfoadenopatie, tumori);
- eseguire biopsia TC-guidata.
- La TC del torace ad alta risoluzione oltre ad avere una maggiore sensibilità rispetto alla semplice radiografia del torace nell’individuare e caratterizzare le alterazioni descritte, permette di definire altri reperti e quadri più specifici, ovvero:
- ispessimento dell’interstizio interlobulare, solitamente di tipo reticolare, con una caratterizzazione molto più dettagliata ed accurata rispetto alla RX;
- aspetto “a vetro smerigliato”, che esprime l’interessamento dell’interstizio parieto-alveolare, caratterizzato da una diffusa e sfumata iperdensità dei campi polmonari. Caratteristica è che sono comunque identificabili i vasi e i bronchi perchè di densità minore rispetto alle aree consolidative vere e proprie;
- immagini a nido d’ape (Honey-combing): questa alterazione si osserva nelle fasi tardive delle malattie fibrotiche-interstiziali e riproduce perfettamente le alterazioni anatomo-patologiche di questo stadio, ovvero una la fibrosi diffusa che determina un quadro reticolare grossolano con cisti aeree che sostituiscono il lobulo;
- distorsione architettonica polmonare: nelle fasi avanzate si associa una fibrosi severa che riduce il volume polmonare;
- bronchiectasie e bronchioloectasie da trazione;
Immagine 01
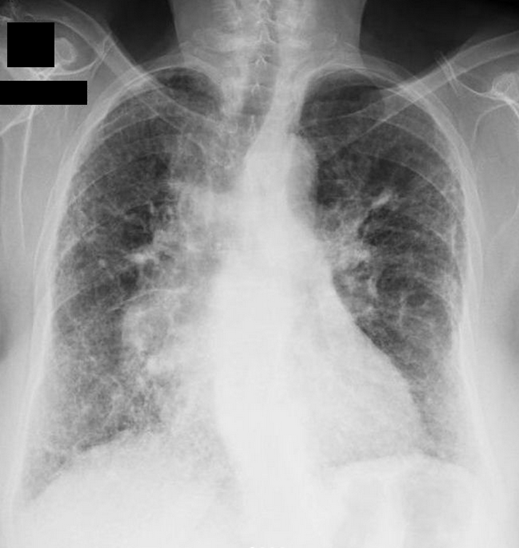
Immagine 01. RX torace mostrante un diffuso ispessimento dell’interstizio polmonare bilaterale peri-bronco-vascolare prevalentemente a sede ilo-perilare ed ilo-basale ed alcune piccole bronchiectasie da trazione.
Immagine 02
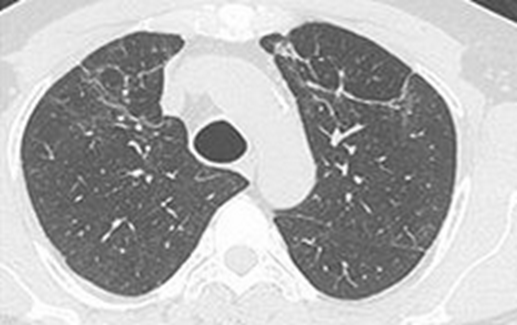
Immagine 02. TC del torace ad alta risoluzione mostrante ispessimento dei setti interlobulari ad entrambi i lobi superiori con evidenza bilateralmente di alcune strie fibrotiche ed a sinistra di alcune piccole bronchiectasie da trazione.
Immagine 03
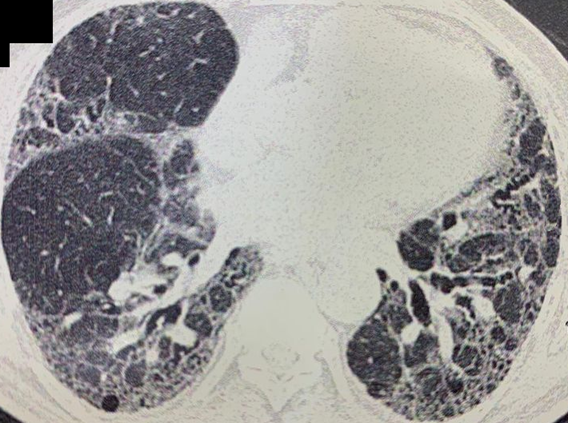
Immagine 03. TC del torace ad alta risoluzione mostrante diffuso ispessimento dell’interstizio prevalentemente interlobulare con evidenza di bronchiectasie da trazione, alcune bolle di enfisema polmonare ed aree di honey-combing.