Dettagli
- Descrizione
- Epidemiologia
- Possibili fattori di rischio
- Eziologia e patogenesi
- Sintomi e EO
- Esame obiettivo
- Diagnosi e stadiazione
- Terapia
- Complicazioni e prognosi
- Prevenzione e screening
- Immagine 01
- Immagine 02
- Immagine 03
- Immagine 04
- Immagine 05
07.15 – [07.06 – Linfomi T] Micosi fungoide
Descrizione
- La sindrome di Sezary è una variante comune e tipicamente indolente di un linfoma cutaneo a cellule T (CTCL) che si presenta principalmente nella pelle, ma può progredire anche nei linfonodi, nel sangue o, meno comunemente, in altri organi.
Tipi (questa parte io la eliminerei, perché l’articolo è incentrato sulla MF e non su sezary)
- I linfomi cutanei primari a cellule T (CTCL) sono un gruppo di linfomi non-Hodgkin che si manifestano primariamente nella pelle ma possono progredire nei linfonodi, nel sangue e negli organi viscerali. Tra i più frequenti troviamo:
- micosi fungoide: è il tipo più comune (60% di tutti i CTCL e quasi il 50% di tutti i linfomi cutanei primari), di natura indolente, che colpisce principalmente la pelle e nasce dalle cellule T di memoria;
- sindrome di Sezary: variante leucemica ed eritrodermica della micosi fungoide, caratterizzata da un significativo coinvolgimento del sangue, con prurito e spesso linfoadenopatia generalizzata; nasce dalle cellule T di memoria del timo e rappresenta il 2% dei casi di CTCL.
- La micosi fungoide si può classificare poi in diversi sottotipi. Quella classica è caratterizzata da grandi chiazze eritematose che possono evolvere in placche, con possibili formazioni tumorali o coinvolgimenti extracutanei; l’istologia mostra linfociti atipici in un infiltrato epidermotropico, con cellule allineate lungo la zona di giunzione e la formazione di ammassi intraepidermici di linfociti atipici (chiamati microascessi di Pautrier). Tra le altre varianti troviamo:
- micosi fungoide follicolotropica (FMF), che si presenta inizialmente con papule follicolari, lesioni acneiformi con comedoni e cisti, chiazze alopeciche e ha lo stesso decorso indolente delle forme classiche di micosi fungoide; le forme più avanzate invece sono caratterizzate da placche alopeciche più spesse e infiltrati linfocitari profondi; hanno un decorso aggressivo e necessitato di una terapia più importante. Si localizza prevalentemente nella regione della testa e del collo e rappresenta circa il 10% dei casi di micosi fungoide. Istologicamente è caratterizzata da infiltrati prevalentemente follicolotropici (esocitosi di cellule tumorali negli epiteli del follicolo pilifero). Dal punto di vista prognostico, ha un rischio più elevato di progressione, rispetto ad altre varianti di micosi fungoide; qualora invece la malattia in fase precoce fosse limitata esclusivamente alla pelle, sembrerebbe avere una buona prognosi. Inoltre, risulta essere meno responsiva alla terapia topica rispetto all’ MF classica;
- granulomatosi con cute lassa: una variante non comune di micosi fungoide caratterizzata dallo sviluppo di pieghe cutanee voluminose, in particolare nelle aree intertriginose;
- reticulosi localizzata pagetoide: una variante non comune di micosi fungoide, con una prognosi eccellente, caratterizzata da una lesione psoriasiforme solitaria, a crescita lenta, spesso situata alle estremità.
Epidemiologia
- Colpisce prevalentemente i pazienti più anziani (età mediana alla diagnosi 55-60 anni), con una maggiore incidenza negli uomini (rapporto M/F 1,66 per la micosi fungoide e 2,11 per la sindrome di Sezary). Gli individui di colore sono più colpiti (rapporto di incidenza 1,44 rispetto ai pazienti bianchi).
- L’incidenza annuale dei linfomi cutanei a cellule T (CTCL) è stata di circa 6,4-9,6 casi per 1.000.000 di individui negli Stati Uniti dal 1973 al 2002. (so che mi avevi detto di togliere i dati se non sono europei, ma era l’unico dato d’incidenza e mi sembrava corretto mantenerlo)
Possibili fattori di rischio
- Tra i possibili fattori di rischio troviamo: storia familiare di mieloma multiplo, eczema, indice di massa corporea ≥ 30 kg/m2, fumo di sigaretta per ≥ 40 anni, consumo di alcol > 24 g/giorno, immunosoppressione e/o terapia immunosoppressiva (come l’HIV, o dopo un trapianto d’organo).
Eziologia e patogenesi
- Dal punto di vista patogenetico, alla base della malattia vi è una stimolazione antigenica cronica, che porta alla trasformazione neoplastica delle cellule T di memoria e al loro successivo accumulo nella cute. Vi sono prove molto limitate che supportino un’eziologia virale alla base della micosi fungoide.
- Possiamo distinguere tre diverse fasi cliniche, che possono verificarsi in modo sequenziale o simultaneo:
- fase a chiazze, caratterizzata da chiazze piane, caratterizzate istologicamente da un infiltrato linfocitario nel derma superficiale;
- fase a placche, in cui si sviluppano placche sottili, che possono fondersi per formare placche più grandi;
- fase tumorale, con lesioni sporgenti e spesso ulceranti caratterizzate da una crescita verticale importante.
- I microascessi di Pautrier sono caratterizzati dal raggruppamento di cellule T clonali intorno a cellule di Langerhans.
Sintomi e EO
- La micosi fungoide classica si presenta in maniera abbastanza stereotipata, con chiazze/placche pruriginose localizzate nelle aree non fotoesposte, principalmente seni, glutei, tronco inferiore e inguine. Raramente si può presentare con lesioni ipopigmentate, più spesso in bambini o adolescenti, o persone di pelle scura. Le forme tumorali o eritrodermiche si verificano in circa il 30% dei pazienti all’inizio della malattia.
- La sindrome di Sezary si presenta inizialmente con un’eritrodermia molto pruriginosa, con o senza linfoadenopatia. Può essere preceduta da sintomi prodromici (prurito o dermatite aspecifica), ma più spesso insorge de novo. Raramente, rappresenta una complicazione della micosi fungoide classica.
Esame obiettivo
- Per il completamento diagnostico, è necessario eseguire un esame obiettivo completo, effettuando valutazioni circa l’’estensione cutanea e la tipologia delle lesioni (chiazza, placca, tumore, eritrodermia), palpazione delle regioni linfonodali periferiche e degli organi profondi per l’individuazione di un eventuale linfadenopatia o organomegalie.
- L’estensione della malattia si valuta determinando la percentuale di superficie corporea colpita (il palmo delle mani più le dita equivalgono all’1% della superficie corporea). Le zone maggiormente colpite sono quelle non fotoesposte: seni, glutei, tronco inferiore e inguine.
- Le lesioni cutanee tipiche, a seconda dello stadio, sono:
- chiazza, di qualsiasi dimensione, senza rilevatezza o indurimento; può essere ipo- o iperpigmentata (soprattutto nei bambini e nei pazienti di pelle scura) e ricoperta da squame o croste;
- placca, rilevata o indurita e può presentarsi con squame, croste, poichilodermia o ulcerazione;
- tumore, lesione solida o nodulare ≥ 1 cm di diametro con evidenza di profondità e/o crescita verticale.
Diagnosi e stadiazione
- La diagnosi è basata principalmente sulla presentazione clinica, istopatologica, immunopatologica e biologico-molecolare della lesione cutanea. La conferma diagnostica si ha comunque con la biopsia. Qualora quest’ultima non fosse diagnostica, bisognerebbe valutare il sangue periferico per la ricerca delle cellule di Sezary, (raccomandata per qualsiasi paziente con malattia T2-T4 o se si sospetta malattia extracutanea) ed effettuare una biopsia dei linfonodi o dei siti extracutanei sospetti.
Panoramica dei test
- Oltre ai test di routine sopradescritti, bisogna effettuare degli esami del sangue, per caratterizzare correttamente la malattia. Tra questi troviamo:
- emocromo completo con determinazione della conta assoluta dei linfociti;
- studi di citometria a flusso per la ricerca delle cellule di Sezary;
- pannello metabolico completo e misurazione dell’enzima lattato deidrogenasi;
- PCR sui linfociti del sangue periferico per la ricerca del riarrangiamento del gene TCR, se si sospetta un coinvolgimento del sangue.
- Si possono effettuare anche studi di imaging se vi sono dati prognostici negativi (≥ T2b, grandi cellule trasformate, follicolotropismo, adenopatia palpabile o risultati di laboratorio anormali), come ad esempio la TC con mdc di torace, addome e regione pelvica o la PET total body.
- Altri test utili includono:
- biopsia dei linfonodi o dei siti extracutanei sospetti (se la biopsia della cute non è diagnostica);
- biopsia del midollo osseo se si sospetta un coinvolgimento del midollo o vi è un’anomalia ematologica atipica;
- test sierologico per la ricerca degli anticorpi contro il virus umano linfotropo delle cellule T di tipo 1, se il paziente è considerato a rischio;
- studi immunoistochimici di campioni bioptici per determinarne l’immunofenotipo.
- Si raccomanda un test di gravidanza nelle donne in età fertile, poiché molte terapie topiche e sistemiche, come i retinoidi, sono controindicate durante la gravidanza.
Sistemi di stadiazione
- Il sistema di stadiazione più usato è il TNMB, che valuta il coinvolgimento cutaneo con “T”, quello linfonodale con “N”, l’interessamento viscerale con “M” e del sangue con “B”, come proposto nello schema seguente.
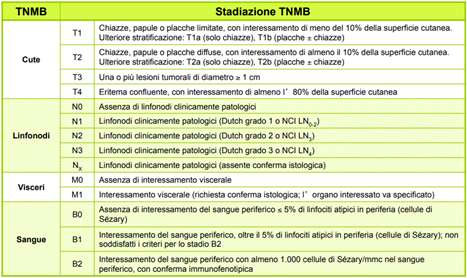
In base ai vari parametri di TNMB, si può calcolare lo stadio della micosi fungoide, come mostrato nella seguente tabella.
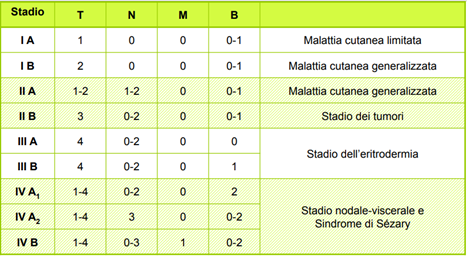
Terapia
- Dato che la maggior parte dei trattamenti non sono risolutivi e i sintomi della malattia hanno spesso un impatto importante sulla qualità della vita, il trattamento ottimale dipende dagli obiettivi individuali del paziente, tenendo conto che bisogna comunque evitare eventuali tossicità dovute al trattamento sistemico. Non vi sono prove di evidenza che dimostrino la reale efficacia della terapia, ma è importante che tutti i pazienti ricevano cure di supporto, per diminuire l’impatto dei sintomi sulla qualità di vita. Nel caso vi fosse una risposta positiva al trattamento, bisognerebbe considerare anche una terapia di mantenimento, per ottimizzare la durata della risposta. Qualora invece vi fosse recidiva o non risposta al trattamento, bisogna considerare l’arruolamento in uno studio clinico o ripetere la biopsia, se si sospetta la trasformazione in un linfoma a grandi cellule.
- Il trattamento varia ovviamente in base allo stadio clinico. Nelle micosi fungoidi in stadio clinico IA (chiazze o placche con coinvolgimento cutaneo <10% e assenza di coinvolgimento extracutaneo) si possono usare:
- corticosteroidi topici ad alta potenza: una o due volte al giorno, meglio se in occlusione; danno una risposta completa nella maggior parte dei casi; in questo caso, non bisogna impostare un regime di mantenimento, poiché un uso reiterato può portare ad atrofia cutanea. Pertanto, è necessario ricominciare la terapia in caso di recidiva della malattia;
- chemioterapici topici, come la mecloretamina (disponibile in gel o unguento): risposta positiva nel 70-80% dei casi, con tempo libero di malattia mediamente di circa 6 mesi; tra gli effetti collaterali più frequenti troviamo una reazione di ipersensibilità ritardata e aree residue di ipo/iperpigmentazione;
- retinoidi topici, utili soprattutto nella variante follicolotropica; possono dare fotosensibilità;
- imiquimod topico al 5%, una volta al giorno per 3 mesi: si tratta di un immunomodulatore, che stimola la produzione locale di citochine infiammatorie e esercita un’azione proapoptotica nei confronti delle cellule tumorali; si utilizza solo come ultima scelta, qualora avessero fallito le altre terapie topiche; può causare una reazione infiammatoria importante, con edema, eritema e ulcerazioni;
- irradiazione con raggi X, poiché l’MF è altamente sensibile alle radiazioni; sfortunatamente, gli effetti collaterali ne limitano l’utilizzo;
- Fototerapia: si usa UVA (320/400 nm) + psoraleni inducenti (PUVA) o UVB a banda stretta (311 nm); dato il rischio di sviluppare melanomi cutanei, la fototerapia è controindicata nei pazienti con anamnesi personale o familiare di tumori cutanei; solitamente si preferiscono gli UVB a banda stretta, per i minori effetti collaterali; si effettua dalle 3 alle 5 volte a settimana con dose sempre crescente.
- Negli stadi IB (chiazze e placche che coinvolgono > 10% della superficie cutanea, senza interessamento extracutaneo) e IIA (qualsiasi grado di coinvolgimento cutaneo e con interessamento linfonodale) si possono utilizzare le seguenti terapie:
- corticosteroidi topici;
- chemioterapia topica con mostarda azotata;
- terapia con fascio di elettroni cutanei totale (TSEBT), alla dose di 30-36 Gy distribuite in 10 settimane;
- Fototerapia: PUVA o UVB, come per lo stadio IA;
- qualora vi fosse un fallimento di una delle terapie sovraproposte, è consigliabile shiftare verso un’altra terapia topica o aggiungerla in combinazione alla precedente.
- Nello stadio IIB (presenza di tumori) localizzato si può utilizzare la stessa terapia utilizzato per lo stadio a chiazze/placche; se invece vi è un coinvolgimento cutaneo esteso si utilizzano terapie sistemiche (agenti biologici, inibitori dell’istone deacetilasi, brentuximab vedotin) da scegliere in base ad eventuali controindicazioni, in aggiunta, ove possibile, alla terapia topica.
- Negli stadi IIIA e IIIB (stadio eritrodermico: coinvolgimento > 80%, senza interessamento extracutaneo) si preferisce l’uso di una terapia sistemica (retinoidi, metotrexato a basso dosaggio, brentuximab vedotin, inibitore dell’istone deacetilasi, alemtuzumab).
- Anche nello stadio IV (coinvolgimento del sangue o degli organi interni) è necessario l’uso della terapia sistemica. Le opzioni terapeutiche sono le stesse viste negli stadi precedenti. In aggiunta, si può optare anche per una fotoferesi extracorporea (ECP), spesso preferita perché non causa immunosoppressione, a differenza di molte altre terapie sistemiche.
Complicazioni e prognosi
- Tra le complicazioni più frequenti vi è il prurito, presente in quasi il 90% dei pazienti con linfoma cutaneo a cellule T, riducendone la qualità di vita. Vi sono comunque diverse opzioni terapeutiche per alleviare questo sintomo, spesso invalidante.
- Le lesioni sono spesso soggette a sovrainfezioni virali, come da herpes simplex virus (HSV) o da herpes zoster virus (HZV), o batteriche da Staphylococcus aureus o bastoncini gram-negativi (spesso nelle lesioni necrotiche), con possibili complicanze settiche. La prevenzione delle infezioni è perciò un aspetto molto importante nella corretta gestione dei pazienti con micosi fungoide.
- Per quanto riguarda la prognosi, la micosi fungoide classica ha un decorso indolente, con lesioni che possono essere stabili per anni o addirittura decenni. Fattori prognostici associati alla progressione della malattia sono: età più avanzata alla presentazione, estensione e tipo di coinvolgimento della pelle (placche o tumori), presenza di malattia extracutanea, grado di coinvolgimento del sangue periferico (definito da misurazioni citometriche delle cellule di Sezary), livelli elevati di lattato deidrogenasi, trasformazione in linfoma a grandi cellule.
- La sopravvivenza mediana per i pazienti con micosi fungoide in una coorte prospettica di 1.263 pazienti (età media 55 anni) è:
- uguale alla popolazione generale per i pazienti con malattia T1-T2a;
- 17 anni per i pazienti con malattia T2b;
- 6 anni per i pazienti con malattia T3;
- 5 anni per i pazienti con malattia T4.
Prevenzione e screening
- Non vi sono metodiche di prevenzione o screening disponibili.
Immagine 01
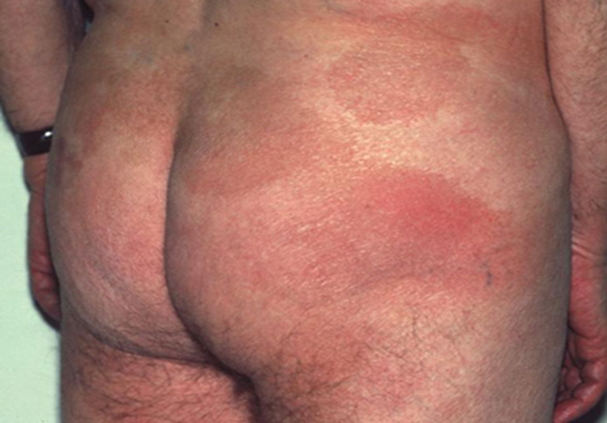
Immagine 01. Micosi fungoide a chiazze: da notare l’interessamento della sede glutea, molto tipica.
Immagine 02
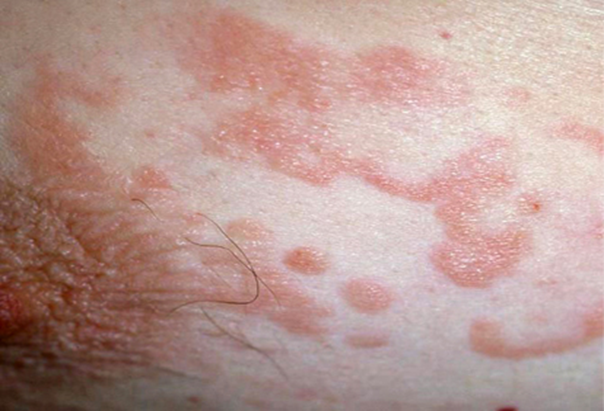
Immagine 02. Micosi fungoide a placche: da notare la rilevatezza e l’aspetto a risoluzione centrale delle lesioni, tipico appunto delle micosi, da cui prende il nome.
Immagine 03
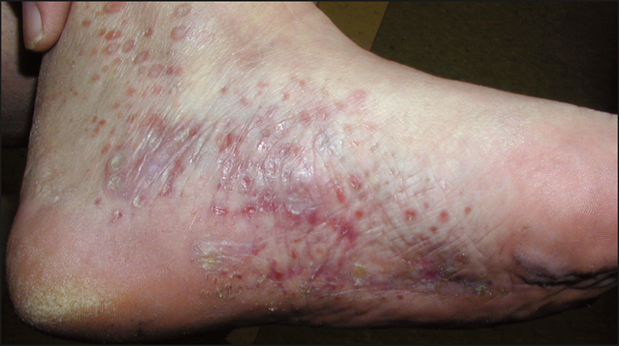
Immagine 03. Micosi fungoide variante follicolotropica in fase iniziale: da notare le papule insorte in corrispondenza degli osti follicolari
Immagine 04
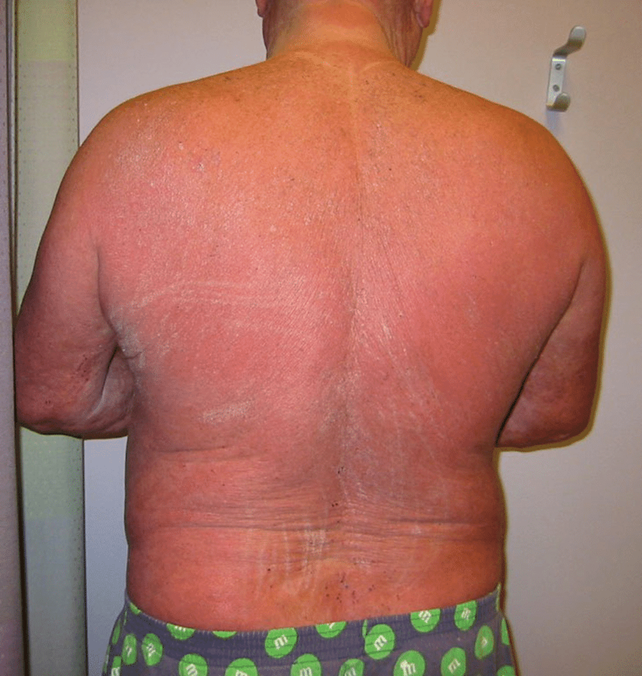
Immagine 04. Sindrome di Sezary: variante eritrodermica della micosi fungoide, a prognosi nettamente peggiore.
Immagine 05
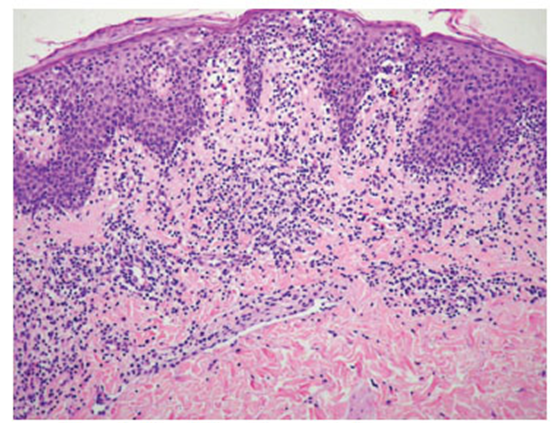
Immagine 05. Micosi fungoide vista all’esame istologico: da notare il pattern psoriasiforme, con infiltrato infiammatorio a banda e modesto epidermotropismo dei linfociti.