Dettagli
- Definizione
- Epidemiologia
- Presentazione clinica
- Patologia
- Caratteristiche radiologiche
- Prognosi
- Trattamento
- Complicanze
- Immagine 01
- Immagine 02
02.38 – [Radiodiagnostica] Sarcoidosi
Definizione
- La sarcoidosi è una patologia infiammatoria granulomatosa multisistemica ad eziologia sconosciuta, caratterizzata dalla presenza di granulomi non caseosi in uno o più organi o tessuti.
- I polmoni e il sistema linfatico sono gli organi colpiti il più delle volte, ma la sarcoidosi può interessare qualsiasi organo e si caratterizza per un’ampia diversità di manifestazioni cliniche e radiologiche.
Epidemiologia
- La sarcoidosi si manifesta in tutte le aree geografiche del mondo, in tutte le età e razze.
- Tuttavia sembra essere più frequente fra la 2° e 4° decade di vita, con una leggera predominanza nel sesso femminile, nella razza afro-americana e nei caucasici dell’Europa settentrionale ed è stato riportato un clustering familiare della sarcoidosi che suggerisce una componente genetica.
Presentazione clinica
- La presentazione clinica è variabile e la diagnosi è di solito fatta sulla combinazione di caratteristiche cliniche e radiologiche:
- metà dei pazienti sono asintomatici;
- il resto sviluppa malattie respiratorie (es. tosse e dispnea) o cutanee (es. eritema nodoso, lupus pernio, cicatrici, placche);
- coinvolgimento delle ghiandole lacrimali e salivari è relativamente comune;
- circa il 5% dei pazienti sviluppa la neurosarcoidosi.
- Inoltre, i pazienti possono avere presentazioni sindromiche specifiche:
- la sindrome di Löfgren: una presentazione acuta non comune ma specifica della sarcoidosi, solitamente autolimitante e con una prognosi migliore, che si presenta clinicamente con triade poliartrite acuta, eritema nodoso e adenopatia ilare e spesso causa febbre, malessere generale, uveite e talvolta interessamento parotideo;
- sindrome di Heerfordt: una variante non comune della sarcoidosi che si manifesta con gonfiore della ghiandola parotide, uveite anteriore, febbre cronica e meno frequentemente paralisi del nervo facciale.
Patologia
- La sarcoidosi è una malattia multisistemica ad eziologia sconosciuta, caratterizzata dalla formazione di granulomi infiammatori non caseosi nei tessuti colpiti.
- Istologicamente, le lesioni mostrano caratteristicamente l’assenza di una componente necrotica, tranne in rari casi (la cosiddetta “granulomatosi sarcoidea necrotizzante”). I granulomi possono risolversi spontaneamente o progredire in fibrosi.
- Si pensa che rappresenti un disordine della risposta immunitaria, in particolare dell’immunità cellulo-mediata.
- Il genoma di microrganismi come il Mycobacterium e propionibacterium sono stati rilevati in alcune lesioni da sarcoidosi, sollevando la possibilità di un trigger infettivo.
- Il clustering familiare riportato in alcuni casi e la presentazione nei gemelli monozigoti suggeriscono che la suscettibilità genetica può giocare un ruolo in tale malattia.
Caratteristiche radiologiche
RX
- Oltre il 90% dei pazienti con sarcoidosi presentano alterazioni toraciche della malattia, in genere visibili con RX del torace.
- Alla radiografia del torace si apprezzerà un ispessimento dell’interstizio polmonare più o meno accentuato con la presenza di uno slargamento ilare bilaterale indice di linfoadenomegalie ilari.
- In effetti, lo “stadio” della sarcoidosi viene ancora oggi convenzionalmente determinato proprio sulla base del RX:
- stadio 0: RX negativo, ma evidenza di malattia con biopsia transbronchiale e/o localizzazioni extratoraciche (cute, occhi, ecc). E’ una evenienza rara;
- stadio I: adenopatie toraciche isolate;
- stadio II: adenopatie associate ad interstiziopatia polmonare;
- stadio III: interstiziopatia polmonare senza adenopatie;
- stadio IV: fibrosi.
- Bisogna precisare che il concetto di questa classificazione, ovvero che la malattia proceda da uno stadio a quello successivo non è del tutto corretta, perché la sarcoidosi può esordire in qualunque stadio; inoltre, un mezzo diagnostico sensibile come la HRCT (TC torace ad alta risoluzione) può dimostrare la presenza di patologia polmonare anche nello stadio I o segni di attività persistente nello stadio IV.
- Tale classificazione ha però un importante valore prognostico.
TC
- La HRCT nella sarcoidosi viene in genere eseguita:
- in casi con presentazione clinico-radiografica atipica;
- per caratterizzare meglio il quadro interstiziale e linfonodale ilo-mediastinico;
- nel sospetto di complicanze;
- per guidare la biopsia broncoscopica.
- La HRCT è più sensibile e specifica del RX e può aiutare a stabilire l‘estensione (gravità) del danno parenchimale, l‘attività della malattia e l‘efficacia della terapia.
- L’interstiziopatia sarcoidea, ben apprezzabile e caratterizzabile con l’HRCT, può avere vari aspetti, ma tipicamente si presenta con ispessimento reticolare o reticolo-micronodulare dell’interstizio polmonare prevalentemente in sede para-ilare, nei lobi superiori e dorsalmente.
- I noduli sono in realtà aggregati di decine o centinaia di granulomi sarcoidei microscopici.
- La HRCT è in grado di evidenziare la classica distribuzione “perilinfatica” delle lesioni, lungo i setti interlobulari, la pleura, a sede intrascissurale e lungo bronchi e vasi, cioè lungo le vie di drenaggio linfatico del polmone. Questo tipo di distribuzione è molto orientativo per la sarcoidosi, ma può essere riscontrato anche in altre malattie, come silicosi, linfangite carcinomatosa, amiloidosi: in questo caso la HRCT, consentendo di riconoscere il cosiddetto pattern di diffusione, indirizza verso la diagnosi corretta o, almeno, riduce notevolmente la gamma delle diagnosi differenziali possibili.
- L’evoluzione fibrotica, quando si verifica, determina opacità retraenti grossolane, strie e reticoli predominanti in regione para-ilare ed ai lobi superiori, bronchiectasie da trazione (fibrotica) ed enfisema para-cicatriziale.
- La HRCT inoltre permette di apprezzare la presenza di linfoadenopatie ilari bilaterali simmetriche e di linfoadenopatie mediastiniche.
- Quella di sarcoidosi è una tipica diagnosi “integrata”, basata cioè sulla compatibilità delle evidenze cliniche, radiologiche e bioptiche e sulla esclusione di altre malattie granulomatose che potrebbero presentarsi in modo simile (principalmente infezioni come TBC, micobatteriosi atipiche, micosi).
Prognosi
- Il coinvolgimento polmonare è responsabile della maggior parte della morbilità e della mortalità nei pazienti con sarcoidosi.
- Il tasso di mortalità complessivo è di circa il 5.
- La probabilità di risoluzione dipende dallo stadio della malattia al momento della presentazione:
- stadio I: 60% di risoluzione spontanea entro 1-2 anni;
- stadio II: 40% (richiedono cicli prolungati di terapia steroidea);
- stadio III: 10% (richiedono cicli prolungati di terapia steroidea);
- stadio IV: lesioni fibrotiche irreversibili.
Trattamento
- Il trattamento è principalmente con l’uso giudizioso di corticosteroidi, per evitare gli effetti avversi dei farmaci.
Complicanze
- Le complicanze dipendono dal modello sistemico di coinvolgimento della malattia. Poiché la sarcoidosi polmonare è di gran lunga la manifestazione più comune, più frequenti saranno le complicanze toraciche, che includono:
- fibrosi polmonare (stadio IV);
- ipertensione arteriosa polmonare e polmonare;
- aspergillomi, che possono essere complicati da emottisi.
Immagine 01
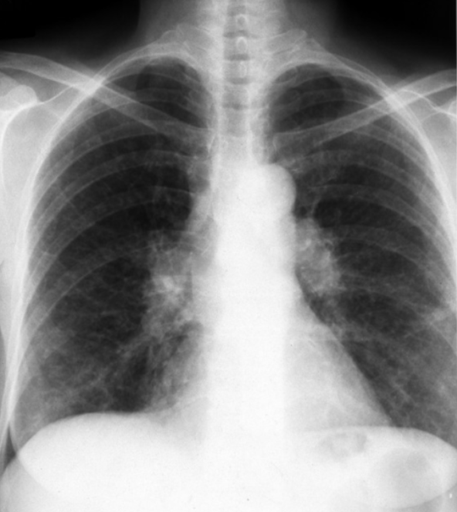
Immagine 01. RX torace mostrante minimo ispessimento dell’interstizio polmonare bilateralmente soprattutto a sede ilo-perilare ed ilo-basale, con associato slargamento ilare bilaterale da linfoadenopatia.
Immagine 02
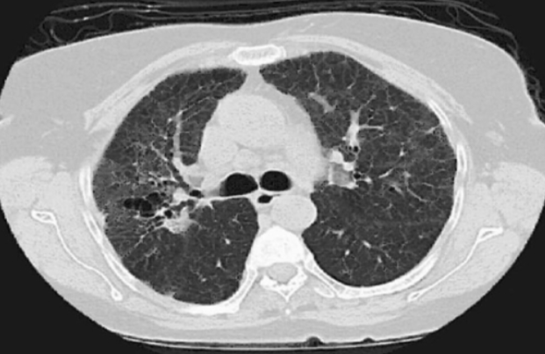
Immagine 02. TC torace mostrante ispessimento dell’interstizio polmonare bilateralmente soprattutto a destra, con pattern fibro-nodulare ed associate bronchiectasie da trazione ed apprezzabilità di linfoadenopatie mediastiniche ed ilari bilaterali.