Dettagli
Classificazione delle patologie interstiziali
- Le “pneumopatie interstiziali idiopatiche” o “malattie interstiziali del polmone (ILD)” o “pneumopatie infiltrative diffuse” rappresentano un gruppo eterogeneo di patologie a interessamento prevalente, ma non esclusivo, dello spazio interstiziale (Immagine 01), ovvero dello spazio compreso tra le membrane basali dell’epitelio e dell’endotelio: questo rappresenta infatti il sito più colpito dalle lesioni anche se queste possono estendersi agli spazi aerei, alle vie aeree periferiche e ai vasi. Esse sono il risultato di un danno del parenchima polmonare da parte di diversi pattern di infiammazione e fibrosi.
- Le malattie interstiziali del polmone riguardano circa il 15% di tutte le malattie respiratorie e quindi sono malattie abbastanza rare. Le differenti entità nosologiche appartenenti a questo gruppo sono però circa 350 e rappresentano pertanto una sfida diagnostica differenziale anche per lo pneumologo più esperto.
- I differenti quadri clinici possono essere raggruppati sotto un denominatore comune poiché l’iter diagnostico è solitamente lo stesso.
- Sono patologie caratterizzate per lo più da un quadro fisiopatologico di tipo restrittivo e da una compromissione della diffusione alveolo-capillare (DLCO); più raramente, in alcune patologie prevalentemente bronchiolo-centriche, può essere osservato invece un quadro di tipo ostruttivo.
- In presenza di un’interstiziopatia polmonare, il primo passo nella diagnosi differenziale è quello di indagare ogni possibile fattore causale, onde discriminare tra quadri a eziologia nota (ad es. da cause occupazionali, o correlati all’inalazione di polveri nell’ambito lavorativo, oppure associati alle connettivopatie, oppure ancora ai farmaci), e quadri a eziologia ignota, ovvero le forme “idiopatiche” (IIP). Le polmoniti interstiziali idiopatiche, oggetto di trattazione in questo capitolo, comprendono quindi un sottogruppo di patologie infiltrative diffuse del polmone, acute o croniche, che originano da un danno parenchimale polmonare e che sono caratterizzate da infiammazione e fibrosi dell’interstizio senza una causa determinante nota.
- La più recente classificazione delle IIP (American Thoracic Society/European Respiratory Society, 2013) prevede una distinzione tra forme comuni, rare e non classificabili (Tabella 01
- Le IIP vengono solitamente distinte dalle altre patologie parenchimali diffuse del polmone sulla base delle caratteristiche cliniche, dell’obiettività, del quadro radiologico e delle caratteristiche istopatologiche, anche se può essere molto difficile distinguerle da altre patologie come l’asbestosi, la polmonite cronica da ipersensibilità, la sarcoidosi e le malattie autoimmuni.
- La tabella 02 riporta la distribuzione percentuale delle differenti forme di IIP.
- Esse possono avere un decorso acuto, sub-acuto o cronico.
- I sintomi sono: la dispnea, presente nelle fasi iniziali solo sotto sforzo e, nelle fasi avanzate, anche a riposo, e la tosse stizzosa.
- L’approccio al paziente con sospetta malattia polmonare parenchimale diffusa (Tabella 03) comincia con l’acquisizione di un’anamnesi completa, di una radiografia del torace e dei test di funzionalità respiratoria. La raccolta anamnestica deve comprendere dati sull’esordio dei sintomi, sulla loro evoluzione e sul decorso clinico, in particolare sull’eventuale presenza di comorbilità; sono anche importanti una valutazione del rischio lavorativo di esposizione a inalanti tossici, la familiarità per neoplasie o per malattie respiratorie, i farmaci assunti. L’esame obiettivo del torace può essere normale oppure possono essere presenti i tipici “crepitii a velcro” che testimoniano l’interessamento interstiziale polmonare.
- Gli esami di laboratorio non sono specifici, ma possono aiutare nella diagnosi differenziale con le altre pneumopatie infiltrative da causa conosciuta (es. dosaggio degli anticorpi anti-nucleo, anti-antigeni nucleari estraibili, anti-citoplasma dei neutrofili, anti-citrullina, fattore reumatoide, ecc.).
- La radiografia del torace è utile per valutare il pattern generale di distribuzione della malattia, anche se manca di specificità e sensibilità: essa correla poco con il pattern istologico, con la distribuzione anatomica e la gravità della malattia. La TC torace ad alta risoluzione (HRCT) ha invece una maggior capacità di visualizzare i dettagli fini ed ha rimpiazzato la radiografia convenzionale del torace nella diagnostica delle IIP; solitamente viene utilizzato un intervallo di scansione di 10 mm. Il potere di risoluzione della HRCT è di 0.2-0.3 mm: la più piccola unità anatomica di polmone visualizzabile alla HRCT è il lobulo polmonare secondario che è la più piccola area di parenchima polmonare circondata da setti di tessuto connettivale. Solitamente i lobuli polmonari secondari sono poco visibili nel polmone sano, ma lo diventano quando i setti sono ispessiti; le arterie centrolobulari del diametro di circa 1 mm possono essere visualizzate, così come le arterie intralobulari che hanno un diametro di circa 0.5 mm; mentre i bronchioli tributari del lobulo secondario non sono visibili in quanto hanno una parete dello spessore di 0.15 mm che è sotto il limite del potere di risoluzione della HRCT. Nella tabella 04 sono riportate le alterazioni radiologiche che si possono osservare nelle IIP: le opacità nodulari, lineari e reticolari, a “vetro
- smerigliato” o ”ground glass”, le lesioni cistiche e i consolidamenti. Nella tabella 05 sono invece riportate le caratteristiche osservabili alla HRCT nelle diverse forme di IIP.
- Le prove respiratorie funzionali dimostrano solitamente una sindrome restrittiva con riduzione dei volumi polmonari e della compliance; i volumi statici sono ridotti in modo non uniforme, la riduzione della capacità vitale è solitamente maggiore di quella del volume residuo e della capacità funzionale residua, per cui la capacità polmonare totale è ridotta in misura minore della capacità vitale e il rapporto RV/TLC può essere aumentato. La ridotta distensibilità polmonare determina volumi correnti piccoli e un’adeguata ventilazione viene mantenuta a spese di un’aumentata frequenza respiratoria. La diffusione (DLCO) è spesso il primo parametro che si riduce. L’emogasanalisi può evidenziare nelle fasi iniziali un’ipossiemia presente solo sotto sforzo e nelle fasi più tardive, anche a riposo. Alcuni parametri funzionali possono essere predittivi di una prognosi sfavorevole: ad es. una ridotta capacità vitale e una ridotta diffusione, oppure il grado di ipossia indotto dall’esercizio.
- Fino a una decina di anni fa si riteneva che la biopsia polmonare rappresentasse il gold standard nella diagnosi delle IIP: tuttavia, anche se la biopsia chirurgica rimane l’esame con maggiore sensibilità diagnostica nelle polmoniti interstiziali, attualmente invece si ritiene utile un approccio dinamico integrato nella diagnosi, che prevede l’interazione tra clinico, radiologo e anatomo-patologo. L’approccio multidisciplinare non sminuisce l’importanza della biopsia polmonare nella diagnosi delle IIP, ma ne definisce i casi di applicabilità, specialmente rispetto alla HRCT che talora può essere già sufficientemente informativa. Le ultime linee guida ATS/ERS del 2013 suggeriscono che la diagnosi delle IIP debba derivare dalla combinazione del pattern istopatologico osservato alla biopsia polmonare con le informazioni cliniche e radiologiche. La biopsia pertanto non è più considerata il gold standard per la diagnosi, ma è parte integrante di un processo di confronto tra clinici, radiologi e patologi.
- L’avvento delle tecniche di chirurgia toracica video-assistita (VATS) ha consentito di effettuare un numero maggiore di biopsie in questa patologia, anche se la biopsia rimane comunque un esame complessivamente poco utilizzato (<15% dei pazienti).
- Nell’esecuzione della biopsia si devono effettuare le seguenti considerazioni: è importante il sito della biopsia, che deve essere effettuata in prossimità delle aree alterate del polmone in modo da includere anche aree di polmone normale ed evitare le aree grossolanamente peggiori alla palpazione; le biopsie devono essere effettuate in più di un lobo polmonare e devono essere profonde in modo da comprendere bene il parenchima sub-pleurico.
- Le biopsie ottenibili per via transbronchiale non sono raccomandabili per la diagnosi di IIP.
- Il lavaggio broncoalveolare (BAL) non sempre è richiesto in questo processo diagnostico, tuttavia può essere utile poichè alcuni profili cellulari correlano con alcuni quadri patologici, per cui un pattern cellulare tipico può confermare una diagnosi clinica; ha un’utilità limitata come indicatore prognostico o come guida alla terapia.
Immagine 01
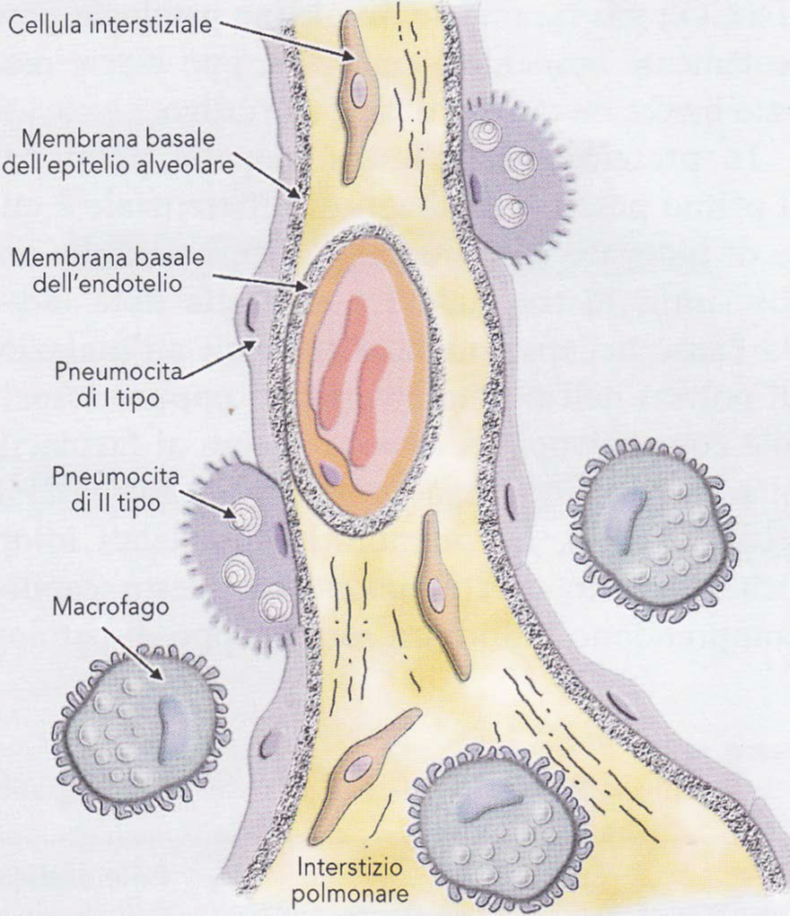
Immagine 01. Interstizio polmonare. L’interstizio polmonare è lo spazio compreso tra l’epitelio alveolare con la sua membrana basale da una parte, e l’endotelio capillare con la sua membrana basale dall’altra, unitamente al connettivo e alle fibre elastiche che costituiscono l’impalcatura di sostegno del parenchima polmonare. Si riconoscono una porzione sottile, dove avviene lo scambio gassoso, e una porzione spessa deputata allo scambio di liquidi e soluti.
Tabella 01

Tabella 01. Classificazione delle pneumopatie interstiziali idiopatiche.
Tabella 02

Tabella 02. Frequenza delle pneumopatie interstiziali idiopatiche.
Tabella 03

Tabella 03. Percorso diagnostico.
Tabella 04

Tabella 04. Alterazioni radiologiche.
Tabella 05
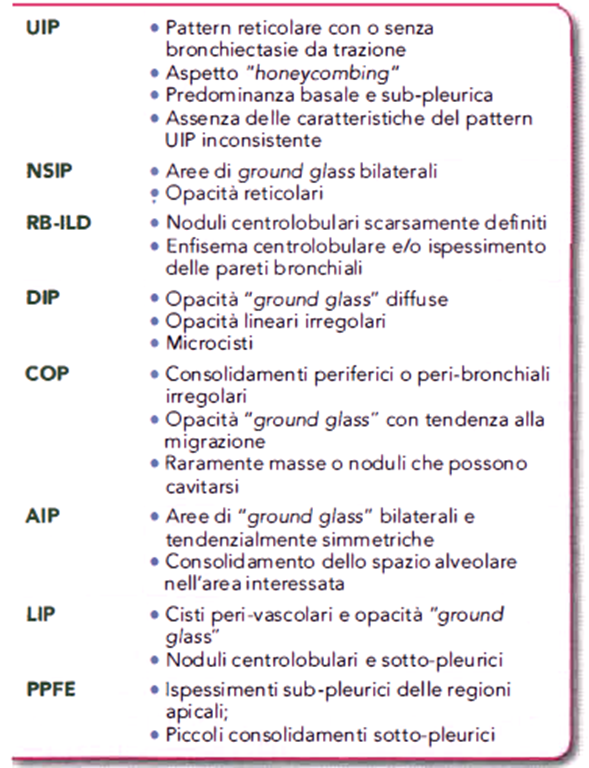
Tabella 05. Caratteristiche alla HRCT.