Dettagli
- Classificazione
- Quadro clinico
- Terapia
Osteogenesi imperfetta
- L’osteogenesi imperfetta (OI) comprende un gruppo eterogeneo di disordini ereditari del tessuto connettivo, caratterizzato da fragilità ossea e altri elementi di disfunzione connettivale. I pazienti affetti sono soggetti a fratture in seguito a traumi di minima entità. Gli altri principali segni clinici sono rappresentati da osteopenia, ipostaturalità, progressive deformità scheletriche, sclere blu, dentinogenesi imperfetta, lassità legamentosa e sordità.
- È una malattia rara con una prevalenza stimata in 0,5-1 caso ogni 10.000 nati.
Classificazione
- Questa malattia è stata classificata in base ad aspetti clinici, radiologici e genetici in quattro sottotipi principali da Sillence (Tabella 01), mentre studi più recenti ne individuano addirittura sette entità distinte. Tuttavia, per un così ampio spettro di variabilità molecolare e di espressività clinica, qualsiasi tentativo di classificazione risulta ancora incompleto o impreciso.
Tabella 01
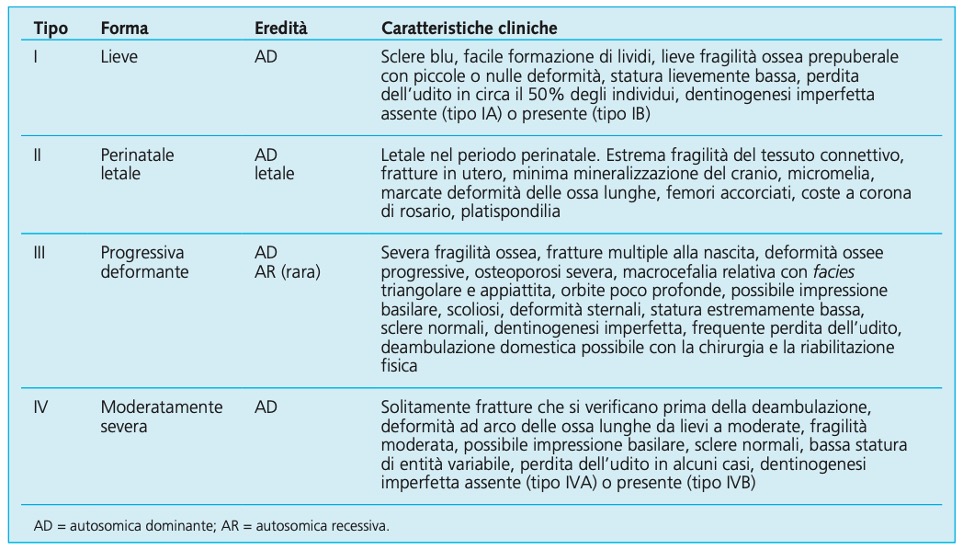
Tabella 01: Classificazione di sillence dell’osteogenesi imperfetta.
Quadro clinico
- Presenta un’ampia eterogeneità anatomo-clinica, che va da forme letali in epoca perinatale (caratterizzate da una statura estremamente bassa e importanti deformità ossee) a quadri in cui è presente solo una fragilità scheletrica con diminuzione della massa ossea, a forme molto lievi che possono eludere l’individuazione clinica.
- Quasi tutti i casi di OI sono causati da una condizione di eterozigosi di mutazioni dominanti a carico di uno dei due geni localizzati sui cromosomi 17 e 7, codificanti rispettivamente per le catene pro-a1 e pro-a2, elementi costitutivi del collagene di tipo I. Questo rappresenta la principale proteina strutturale del tessuto connettivo, in particolare della matrice extracellulare dell’osso, della cute, dei tendini e dei denti.
- Alcune mutazioni danno luogo alla formazione di un collagene di tipo I qualitativamente alterato, evenienza in relazione con i casi più gravi di OI; altre invece non modificano la struttura del collagene di tipo I, ma ne riducono la sintesi, dando luogo a un difetto di tipo quantitativo.
Terapia
- Il trattamento per le forme lievi mira a educare il paziente ad assumere abitudini che gli garantiscano una qualità di vita normale, mentre per le forme più gravi vengono praticati interventi chirurgici con infibuli endomidollari (tradizionali, telescopici o elastici) per ridurre il rischio di frattura e consentire una normale vita di relazione.