Dettagli
- Esostosi osteo-cartilaginea
- Quadro clinico
- Diagnostica per immagini
- Anatomia patologica
- Terapia
- Condroma
- Quadro clinico
- Diagnostica per immagini
- Anatomia patologica
- Terapia
- Condroblastoma
- Osteoma osteoide
- Quadro clinico
- Diagnostica per immagini
- Anatomia patologica
- Terapia
- Tumore gigantocellulare
- Quadro clinico
- Diagnostica per immagini
- Anatomia patologica
- Terapia
- Fibroma istiocitico
Tumori ossei benigni
Esostosi osteo-cartilaginea
- L’esostosi osteo-cartilaginea, detta anche osteocondroma, è uno dei tumori benigni più frequenti dell’apparato scheletrico. È un amartoma, che origina da un’isola ectopica sottoperiostea di cartilagine di coniugazione, la quale si ingrandisce durante il periodo di accrescimento e matura secondo la normale ossificazione encondrale.
- Pur essendo congenita, l’esostosi si manifesta prevalentemente in una fascia di età compresa tra i 10 e i 20 anni, con una predilezione per il sesso maschile.
Si localizza quasi sempre nella metafisi di un osso lungo, più spesso in prossimità delle cartilagini di accrescimento più fertili (femore distale, tibia e omero prossimale), tendendo a portarsi in sede diafisaria con il progredire della crescita corporea. La scapola e l’ileo sono le sedi più comuni di esostosi tra le ossa piatte. La presenza di esostosi multiple è una condizione ereditaria nella maggior parte dei pazienti ed è nota come malattia di Ombredanne o malattia delle esostosi multiple. Tale quadro morboso ha un’incidenza circa 10 volte inferiore rispetto all’esostosi solitaria.
Quadro clinico
- L’esostosi è di regola asintomatica e la diagnosi viene posta per la comparsa di una tumefazione a lento accrescimento. La compressione su strutture circostanti può causare l’insorgenza di dolore su base irritativa. In caso di esostosi peduncolata, è possibile la frattura del peduncolo con comparsa improvvisa di dolore. Obiettivamente si apprezza una massa dura, solidale sul piano osseo e non dolente alla palpazione.
- Poiché le esostosi riducono il potenziale di crescita delle cartilagini di coniugazione, nelle forme generalizzate più gravi si possono osservare accorciamenti (di solito inferiori a 2 cm) e deformità degli arti (Figura 01). Una vecchia esostosi che riprende a crescere in età adulta e si manifesta con dolore deve essere considerata con molta attenzione; la frequenza di trasformazione sarcomatosa è molto bassa in caso di esostosi solitarie, mentre raggiunge il 25% nelle esostosi multiple.
Figura 01
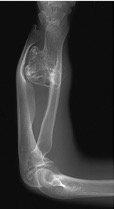
Figura 01: Nella malattia delle esostosi multiple si possono osservare deformità a carico degli arti, in questo caso dell’avambraccio.
Diagnostica per immagini
- Per la diagnosi è sufficiente l’esame radiografico; in rari casi possono essere d’ausilio la TC o la RM per chiarire i rapporti della neoformazione con i tessuti circostanti. Sui radiogrammi è possibile apprezzare la continuità dell’osso corticale e spongioso tra la metafisi e l’esostosi. All’interno della neoformazione possono essere presenti zone iperdiafane o noduli radiopachi; questi ultimi sono dovuti alla calcificazione di porzioni cartilaginee.
Anatomia patologica
- Macroscopicamente si possono osservare esostosi peduncolate (Figura 02), con base d’impianto stretta, oppure sessili, a base larga (Figura 03).
L’apice dell’esostosi, di dimensioni assai variabili, è formato da tessuto cartilagineo normale, che ha maggiore spessore e presenta le caratteristiche della cartilagine di coniugazione nel bambino, per poi assottigliarsi progressivamente, assumendo in età adulta una struttura più simile a quella della cartilagine articolare. Il periostio della metafisi continua sull’esostosi fino al margine con il cappuccio cartilagineo; le trabecole ossee contengono normale tessuto midollare.
La crescita dell’osteocondroma prosegue fino al raggiungimento della maturità scheletrica per poi arre- starsi. Una ripresa della crescita in età adulta deve fare sospettare la trasformazione sarcomatosa (condrosarcoma periferico) dell’esostosi.
Figura 02

Figura 02: Esostosi peduncolata del corpo della scapola. La radiografia in proiezione laterale permette di apprezzare il conflitto della neoformazione con la parete toracica posteriore.
Figura 03

Figura 03: Voluminosa esostosi sessile dell’omero prossimale.
Terapia
- La terapia delle esostosi sintomatiche o sospette per trasformazione maligna è chirurgica e consiste nella loro asportazione, avendo cura di rimuovere anche la base d’impianto per prevenire possibili recidive.
Condroma
- Il condroma, detto anche encondroma, è una neoformazione benigna costituita da cartilagine ialina ben differenziata; si ritiene che sia un amartoma, originato da residui di cartilagine embrionale rimasti inclusi nel tessuto osseo.
- È frequente (rappresenta circa il 10% dei tumori primitivi dell’osso), non presenta predilezione di sesso e si riscontra in tutte le età tra i 10 e i 50 anni.
Il condroma può insorgere in tutte le ossa formatesi su un modello cartilagineo (ossificazione encondrale), ma nel 50% dei casi si localizza alle ossa lunghe delle mani, seguite da quelle del piede. È possibile che l’incidenza in altre sedi possa essere sottostimata per l’assenza di espressività clinica.
Oltre al condroma solitario, che rappresenta la manifestazione più frequente di questa lesione, esiste anche una forma a localizzazioni multiple, nota come encondromatosi o malattia di Ollier. L’associazione di condromi multipli e di angiomi dei tessuti molli è conosciuta come sindrome di Maffucci.
Quadro clinico
- Il condroma resta asintomatico a lungo, ma quando arriva a soffiare la corticale può causare tumefazione e deformità del segmento interessato, soprattutto se superficiale. La comparsa di dolore è di solito associata a una frattura patologica, ma può essere indice di una trasformazione sarcomatosa.
Diagnostica per immagini
- Radiograficamente si apprezza un’area osteolitica centrale, talvolta eccentrica o intracorticale, che può occupare l’intera sezione dell’osso (Figura 04). L’osteolisi ha margini arrotondati e ben definiti, contiene al suo interno calcificazioni in forma di opacità granulari e soffia la corticale diafisaria senza interromperla.
Figura 04

Figura 04: Condroma della falange intermedia del terzo dito.
Anatomia patologica
- Macroscopicamente il condroma appare formato da lobuli di cartilagine ialina, all’interno della quale possono essere presenti zone di calcificazione e necrosi. L’aspetto microscopico è simile a quello della cartilagine normale, con condrociti sparsi o riuniti in gruppi isogeni. Le cellule raramente sono binucleate; i nuclei presentano dimensioni uniformi e di rado sono aumentati di volume.
- La diagnosi differenziale va posta essenzialmente con il condrosarcoma di grado I, per il quale l’istologia non sempre risulta discriminante, mentre altri tre elementi possono essere d’aiuto: la localizzazione (il condrosarcoma è raro nelle ossa della mano), le dimensioni (un diametro > 4 cm è indicativo di condrosarcoma) e la clinica (il condroma è asintomatico in assenza di fratture).
Terapia
- La terapia prevede lo svuotamento accurato della cavità e il suo borraggio (ovvero riempimento) con innesto osseo. Le recidive sono possibili per il permanere di isole cartilaginee.
Condroblastoma
- È un raro tumore benigno di origine cartilaginea, che si manifesta nella maggior parte dei casi in età̀ compresa tra i 10 e i 20 anni, prediligendo il sesso maschile. Ha una spiccata tendenza a localizzarsi in sede epifisaria, sebbene possa superare la cartilagine di accresci- mento e interessare la metafisi. Le epifisi più colpite sono quella distale del femore e quella prossimale di tibia e omero.
- I sintomi sono tardivi e riferiti all’articolazione contigua, con dolenzia, rigidità, tumefazione, ipotrofia muscolare e talvolta versamento.
La radiografia mostra un’area osteolitica epifisaria di dimensioni variabili, a contorni netti e con possibili tenui calcificazioni al suo interno, meglio evidenziate dalla TC o dalla RM (Figura 05). La terapia consiste nell’asportazione della lesione e nel borraggio con innesto osseo, evitando la contaminazione intrarticolare. In alcuni casi (lesioni con caratteristiche di aggressività, recidive) può rendersi necessaria la resezione ossea.
Figura 05
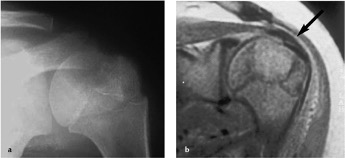
Figura 05: Condroblastoma epifisario dell’omero prossimale. La radiografia mostra un’area osteolitica che raggiunge il profilo superiore della testa omerale (a); la RM permette di apprezzare l’estensione del tumore fino alla cartilagine articolare, che appare lievemente infossata in sede superiore () (b).
Osteoma osteoide
- È un tumore benigno relativamente frequente (3% di tutti i tumori ossei primitivi), il terzo per ordine di incidenza dopo l’esostosi e il fibroma istiocitico. È tipico dell’età giovanile e si osserva solo in modo sporadico prima dei 5 anni e dopo i 30; predilige il sesso maschile.
- La sua localizzazione caratteristica è la diafisi o meta- diafisi delle ossa lunghe degli arti (nell’ordine, femore, tibia e omero), dove si sviluppa all’interno della corti- cale o nella spongiosa in posizione eccentrica. Oltre alle sedi sopracitate, tra le quali va ricordato in partico- lare il femore prossimale, questo tumore può essere osservato nelle ossa della mano e del piede e nell’arco posteriore delle vertebre.
Quadro clinico
- Caratteristica clinica peculiare dell’osteoma osteoide è la precoce e intensa sintomatologia dolorosa, che invece è di regola tardiva nelle altre neoplasie scheletriche. Il dolore è indipendente dall’attività, si accentua durante il riposo notturno e dopo assunzione di sostanze vasodilatatrici, come l’alcol; l’assunzione di salicilati o di altri farmaci antinfiammatori non steroidei fa recedere la sintomatologia. La localizzazione vertebrale può simulare un’irritazione radicolare, con scoliosi antalgica e rigidità.
Diagnostica per immagini
- L’immagine radiografica tipica è quella di una piccola area osteolitica rotondeggiante (1-2 cm di diametro) circondata da un orletto sclerotico, il cosiddetto nidus, all’interno del quale si possono osservare calcificazioni puntiformi. In sede diafisaria il nidus si accompagna a una reazione periostale anche intensa (Figura 06a), che tuttavia non è presente nelle lesioni intracapsulari, come per esempio nel collo del femore.
- La sclerosi periferica può essere tale da mascherare il nidus e rendere necessario il ricorso alla TC (Figura 06b). In casi di difficile localizzazione (rachide) la scintigrafia ossea evidenzia un’intensa ipercaptazione. Problemi di diagnosi differenziale possono nascere nei confronti di alcune osteomieliti croniche (ascesso di Brodie).
Figura 06
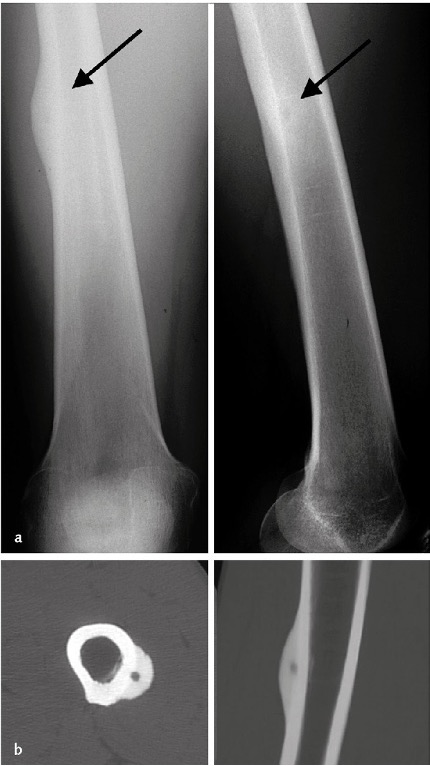
Figura 06: Osteoma osteoide della diafisi femorale. La radiografia mostra l’intensa reazione periostale che maschera la presenza del nidus (a). La TC (sezione assiale e ricostruzione coronale) permette di apprezzare l’osteolisi centrale, circondata dall’osso sclerotico (b).
Anatomia patologica
- L’osteoma osteoide si presenta come una piccola neo-formazione rotondeggiante e iperemica, più soffice rispetto all’osso che la circonda e stridente al taglio. Istologicamente il tessuto tumorale è formato da trabecole di osso immaturo fittamente stipate, tra le quali si sviluppa una rete di capillari dilatati. Sono presenti numerosi osteoblasti e osteoclasti, secondo il normale quadro del rimodellamento osseo, accanto a fibroblasti e fibre nervose. Il tessuto osteoide è più maturo al centro, la parte più vecchia del tumore, dove le trabecole confluiscono in ammassi più compatti, più calcificati e più poveri di cellule.
Terapia
- La terapia classica in passato consisteva nell’asportazione in blocco del nidus e dell’orletto sclerotico circostante, un intervento che spesso comportava esposizioni chirurgiche ampie per raggiungere la lesione. Oggi l’ablazione con radiofrequenze TC guidata viene considerata la terapia di prima scelta per la cura dell’osteoma osteoide.
Tumore gigantocellulare
- Il tumore gigantocellulare (TGC), chiamato anche osteoclastoma, è una neoplasia endomidollare costituita da cellule mononucleate e da cellule giganti multinucleate simil-osteoclastiche, con un potenziale di crescita variabile e imprevedibile. È considerato una neoplasia benigna, quantunque in rari casi possa produrre metastasi polmonari, che peraltro conservano caratteristiche citologiche di benignità.
- Il TGC è frequente (20% di tutti i tumori benigni dell’osso) e colpisce soprattutto giovani adulti, con circa 2/3 dei casi tra i 20 e i 40 anni; l’incidenza nei due sessi è equivalente.
- Si localizza tipicamente nelle metaepifisi delle ossa lunghe, in tre sedi preferenziali: femore distale, tibia prossimale e radio distale. Se l’insorgenza della neoplasia precede la chiusura della fisi, si può osservare la rara localizzazione metafisaria pura.
Quadro clinico
- I sintomi (dolore, limitazione funzionale) sono aspecifici e spesso attribuiti all’articolazione limitrofa, sebbene un versamento articolare sia un’evenienza rara. In fase più avanzata si rileva una tumefazione dovuta alla deformazione dell’osso, a cui si può associare un’accentuazione del reticolo venoso superficiale con cute tesa e termotatto positivo.
Diagnostica per immagini
- L’esame radiografico dimostra una lesione osteolitica uni- o pluricamerata, a limiti sfumati. La corticale appare soffiata e assottigliata, talvolta interrotta, senza evidenza di reazione periostale. L’osteolisi si estende fino all’osso subcondrale, risparmiando la cartilagine articolare.
- In base alle caratteristiche radiografiche, il TGC è stato distinto in tre varianti con importanti risvolti dal punto di vista prognostico e terapeutico:
- calma: corticale integra o poco assottigliata, osteolisi a limiti netti senza estensione all’osso subcondrale;
- attiva: corticale erosa, senza estensione del tumore ai tessuti molli;
- aggressiva: interruzione della corticale con estensione del TGC ai tessuti molli, l’osteolisi raggiunge la cartilagine articolare (Figura 07).
Figura 07
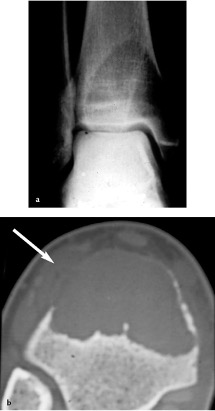
Figura 07: Tumore gigantocellulare della tibia distale (variante aggressiva). La rx dimostra l’estensione dell’osteolisi fino all’osso subcondrale; non si rileva alcuna reazione periostale (a). Quadro TC che mostra l’interruzione della corticale tibiale esterna () con sviluppo extrascheletrico del TGC (b).
Anatomia patologica
- Il TGC si presenta come una massa di consistenza molle e di colore rosso bruno con aree necrotiche ed emorragiche, a limiti netti, ma non delimitati da una capsula. Istologicamente è costituito da cellule mononucleate stromali di forma ovale o fusata, che rappresentano l’elemento neoplastico e appaiono in fase mitotica, frammiste a cellule giganti multinucleate (osteoclasti).
- La classificazione in gradi istologici, proposta in passato, viene oggi ritenuta priva di valore per un giudizio prognostico. L’unica distinzione importante dal punto di vista istologico è quella tra TGC benigno e sarcoma gigantocellulare. Quest’ultimo, come forma maligna ab initio, dimostra peraltro di appartenere più spesso alle categorie degli osteosarcomi, dei fibrosarcomi o degli istiocitomi fibrosi maligni. È invece possi- bile una trasformazione sarcomatosa del TGC benigno, soprattutto dopo terapia radiante.
- La diagnosi differenziale del TGC va posta con diverse neoplasie, ma in particolare con la cisti aneurismatica.
Terapia
- La terapia del TGC va modulata sulla base della classificazione radiografica.
Nella variante calma e in alcune forme attive è sufficiente lo svuotamento accurato della lesione, il trattamento delle pareti della cavità con adiuvanti (bisturi elettrico, fenolo) per eradicare il focolaio neoplastico e il borraggio con innesto osseo. L’incidenza di recidiva con questo protocollo si aggira intorno al 10%. Nelle forme più aggressive si deve ricorrere alla resezione ossea segmentaria, seguita da procedure ricostruttive osteo-articolari.
Fibroma istiocitico
- Il fibroma istiocitico, o non ossificante, è un tumore benigno molto frequente, probabilmente presente in circa 1/3 dei bambini tra i 4 e i 10 anni, anche se la sua reale incidenza non è valutabile per la completa asintomaticità. È un amartoma metafisario composto da istio-fibroblasti, raramente osservabile dopo i 20 anni in virtù della tendenza a una regressione spontanea, soprattutto se di piccole dimensioni. La grande maggioranza dei fibromi istiocitici si localizza in prossimità del ginocchio.
- L’aspetto radiografico è così caratteristico che la diagnosi non necessita di nessun altro accertamento. La lesione si presenta come un piccolo difetto osteolitico, metafisario, eccentrico e superficiale. Ha il maggiore asse orientato secondo la lunghezza dell’osso e forma policiclica, sempre circondato da un orletto sottile di osteosclerosi (Figura 08).
- Non è necessaria alcuna terapia, poiché il fibroma istiocitico arresta la sua crescita con la pubertà tendendo poi a ossificare.
Figura 08
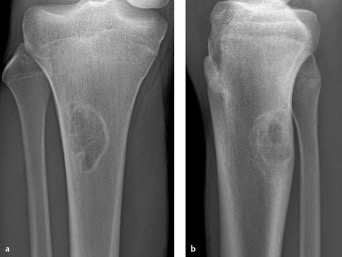
Figura 08: Fibroma istiocitico alla diafisi prossimale di tibia in un ragazzo di 17 anni. Il semplice esame radiografico in proiezione antero-posteriore (a) e laterale (b) permette di porre la diagnosi in virtù dell’aspetto caratteristico della lesione (si veda il testo).