Dettagli
- Condrosarcoma
- Quadro clinico
- Diagnostica per immagini
- Anatomia patologica
- Terapia
- Osteosarcoma
- Quadro clinico
- Diagnostica per immagini
- Anatomia patologica
- Osteosarcomi meno frequenti
- Terapia
- Sarcoma DI ewing
- Quadro clinico
- Diagnostica per immagini
- Anatomia patologica
- Terapia
- Istiocitoma fibroso maligno
- Quadro clinico
- Diagnostica per immagini
- Anatomia patologica
- Terapia
Tumori ossei maligni
Condrosarcoma
- Il condrosarcoma è un tumore maligno caratterizzato dalla produzione, da parte delle cellule neoplastiche, di cartilagine ma non di tessuto osseo.
È il secondo sarcoma dello scheletro per frequenza dopo l’osteosarcoma; interessa l’età adulta (30-60 anni) ed è più frequente nel sesso maschile. Si localizza con maggiore frequenza al bacino, alla scapola e alle metafisi delle ossa lunghe (femore prossimale e distale, tibia e omero prossimali). - I condrosarcomi possono essere distinti:
- a seconda dell’origine, in primitivi e secondari (insorti su preesistenti tumori cartilaginei benigni come esostosi o condromi, soprattutto in caso di malattia di Ombredanne o di Ollier);
- in base alla localizzazione nell’osso, in centrali, periferici e periostei (distinzione che ha un significato pressoché esclusivo nelle ossa lunghe).
- Le possibili varietà di condrosarcoma, tenendo presente che le forme convenzionali sono considerate quella centrale, periferica e periostea, mentre i rimanenti quadri (a cellule chiare, mesenchimale, sdifferenziato) sono considerati varietà del condrosarcoma.
Quadro clinico
- La caratteristica dei condrosarcomi è di avere una crescita molto lenta, con sintomi e segni aspecifici (dolenzia, tumefazione a scarsa evolutività) che spesso durano anni prima che il paziente si rivolga al medico per eseguire degli accertamenti.
Diagnostica per immagini
- Radiograficamente il condrosarcoma centrale è caratterizzato da un’area osteolitica policiclica, con limiti mal definiti verso gli spazi midollari; la corticale appare erosa o interrotta, con scarsa reazione periostale.
- Il condrosarcoma periferico origina solitamente da un’esostosi che si espande nelle parti molli (è importante il dato clinico di “riattivazione” dell’osteocondroma): può essere scarsamente visibile (delineato solo da piccole aree calcificate) (Figura 01) o al contrario essere estesamente radiopaco con aspetto “a cavolfiore” (Figura 02).
- Nel condrosarcoma periosteo, tipicamente radiotrasparente e a sviluppo nelle parti molli, si può osservare una reazione periostale che circonda la base del tumore.
Le metastasi (polmonari) sono piuttosto rare e tardive, la loro comparsa è correlata al grado istologico del tumore.
Figura 01
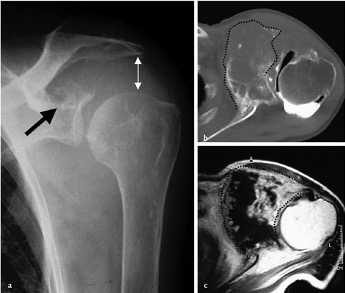
Figura 01: Condrosarcoma di grado III della scapola sinistra. La radiografia permette di apprezzare l’alterazione strutturale del collo scapolare con un’apparente frattura del polo superiore della glena (→). Sono presenti tenui calcificazioni nello spazio subacromiale che appare particolarmente ampio (↔) (a). Sezione assiale TC che consente di apprezzare l’estensione della neoformazione nelle parti molli periscapolari (linea tratteggiata) e l’infiltrazione neo-plastica del collo della scapola (b). Corrispondente sezione RM che definisce in maniera più dettagliata l’estensione del tumore nelle parti molli (linea tratteggiata) (c).
Figura 02

Figura 02: Condrosarcoma periferico (↖) che origina dalla testa e dal collo femorale.
Anatomia patologica
- Il condrosarcoma centrale si presenta sotto forma di noduli, biancastri o grigiastri, che invadono il canale midollare ed erodono la corticale fino a superarla. Quello periferico si sviluppa sulla superficie dell’osso e si espande direttamente nelle parti molli; la sede corticale di impianto è di regola normale. Il condrosarcoma periosteo (o iuxtacorticale) è costituito da una massa di origine periostea che può causare un’erosione “a scodella” della corticale con cui è a contatto.
- L’aspetto microscopico varia da tipo a tipo, con la caratteristica comune della produzione di matrice condroide; possono essere presenti aree di calcificazione, emorragiche e necrotiche più o meno estese all’interno della massa neoplastica.
- La gradazione istologica del condrosarcoma convenzionale si basa sul livello di differenziazione cellulare, sull’entità delle anomalie nucleari e sull’aspetto della sostanza intercellulare.
- È correlata con il decorso e la prognosi della neoplasia e consente di distinguere:
- condrosarcomi ben differenziati, di grado I;
- condrosarcomi differenziati, di grado II;
- condrosarcomi scarsamente differenziati, di grado III.
- Oltre alla cosiddetta “progressione di malignità”, talvolta può verificarsi la comparsa, su un preesistente condrosarcoma centrale di grado I o II, di un tumore più aggressivo (in genere fibrosarcoma, istiocitoma fibroso maligno oppure osteosarcoma). In questo caso si parla di condrosarcoma sdifferenziato.
- Il condrosarcoma mesenchimale è caratterizzato dalla presenza di aree molto indifferenziate, mentre quello a cellule chiare, rarissimo e a prognosi più favorevole, si localizza in sede epifisaria.
Terapia
- La terapia è chirurgica e prevede la resezione ampia della lesione. Lo svuotamento non è indicato perché non ha quasi mai successo, qualunque sia il grado di malignità istologica. Anche la radioterapia e la chemioterapia sono controindicate.
- Le recidive locali si osservano più spesso nelle localizzazioni al tronco piuttosto che agli arti; inoltre, la loro comparsa è quasi certa in caso di trattamento chirurgico inadeguato. Pertanto, la prognosi quoad vitam è influenzata dalla sede del tumore, dal grado istologico e dal trattamento iniziale.
Osteosarcoma
- Dopo il plasmocitoma, l’osteosarcoma è il tumore maligno primitivo dello scheletro più frequente, caratterizzato dalla produzione di sostanza osteoide e/o ossea da parte delle cellule neoplastiche.
Se ne distinguono diverse varietà, con aspetti anatomo-clinici e prognostico-terapeutici specifici. L’osteosarcoma classico rappresenta circa l’80% di tutti i casi. La neoplasia insorge di regola in ossa non colpite da precedenti lesioni; tuttavia, si possono osservare osteosarcomi secondari a malattia di Paget (la forma più comune in età avanzata), a osteomielite cronica e a terapia radiante. Inoltre, un osteosarcoma può insorgere, per progressione di malignità, da un condrosarcoma centrale (condrosarcoma sdifferenziato). - L’incidenza assoluta dell’osteosarcoma è bassa − 2 casi per milione di abitanti per anno (circa 100 casi all’anno in Italia) − rappresentando lo 0,2% di tutte le neoplasie maligne. Colpisce in modo prevalente (75%) soggetti tra i 10 e i 30 anni di età, con una predilezione per il sesso maschile (2:1).
- Pur potendosi sviluppare in qualsiasi distretto scheletrico, la localizzazione più tipica è quella metafisaria delle ossa lunghe: il 70% dei casi è osservato a livello del ginocchio (femore distale e tibia prossimale) e della spalla (omero prossimale).
Quadro clinico
- Il dolore è il più frequente sintomo d’esordio, anche se tardivo. Talvolta riferito a un trauma, si manifesta anche a riposo e tende ad aggravarsi abbastanza rapidamente, con la comparsa di una tumefazione locale (tanto più precoce quanto più superficiale è la localizzazione del tumore) (Figura 03a). L’arrossamento della cute sovrastante, il termotatto positivo e l’eventuale limitazione articolare con versamento sono segni tardivi. Le fratture patologiche sono rare e si osservano nelle forme osteolitiche, aggravandone la prognosi.
Figura 03
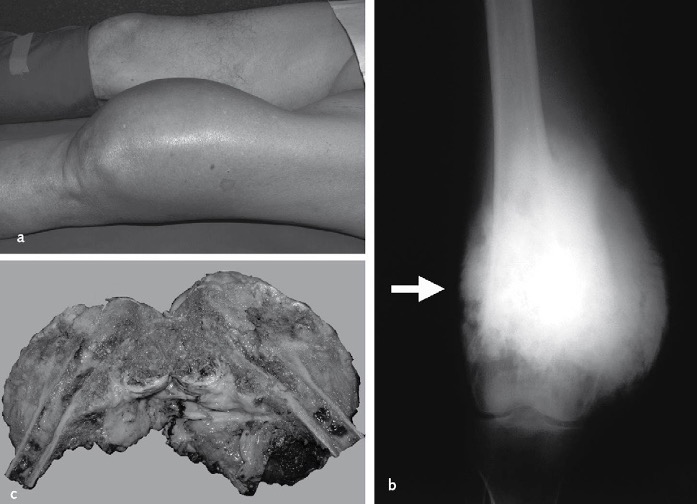
Figura 03: Osteosarcoma del femore distale sinistro. Marcata tumefazione del ginocchio per la progressione avanzata della neoplasia (a). Quadro radiografico che mostra la neo-formazione ossea nei tessuti molli perischeletrici, con aspetti localizzati “a sole radiante” (→) (b). Sezione longitudinale del pezzo operatorio dopo amputazione: la massa neoplastica appare polimorfa per la presenza di zone con caratteristiche istologiche diverse, alternate ad aree emorragiche e necrotiche (c).
Diagnostica per immagini
- Gli esami di laboratorio mostrano un’elevazione della fosfatasi alcalina in circa la metà dei pazienti.
Il quadro radiografico è spesso diagnostico o comunque fortemente indicativo di osteosarcoma. Nella zona colpita si osserva la sovrapposizione di tre aspetti:- la distruzione del preesistente osso corticale e spongioso, con invasione dei tessuti molli;
- la calcificazione e la neoproduzione ossea;
- la reazione periostale. Quest’ultima può assumere i caratteristici aspetti a bulbo di cipolla (ripetute formazioni di strati ossei separati da aree non calcificate, un aspetto considerato tipico del sarcoma di Ewing) e a sole radiante (per formazione di spicule ossee perpendicolari alla corticale) (Figura 03b). Si può inoltre osservare il cosiddetto “triangolo di Codman” (area di distacco del periostio dalla corticale metafisaria, dovuta alla formazione di osso neoplastico a forma di cuneo al di sotto di essa) (Figura 04).
- Questi quadri possono combinarsi tra loro in modo vario e ogni singolo caso può presentare immagini particolari (prevalenza di osteosclerosi o di osteolisi, assenza di reazione periostale).
- La TC può fornire ulteriori informazioni sui caratteri dell’osteolisi e della neoformazione ossea, mentre la RM sull’invasione delle parti molli, sulla vascolarizzazione e sulla presenza di metastasi a distanza nel canale midollare (“skip” metastases). La scintigrafia evidenzia una franca ipercaptazione in sede tumorale, mentre l’arteriografia permette di approfondire il tipo di vascolarizzazione nel planning preoperatorio. L’osteosarcoma classico ha in genere un decorso rapido: metastatizza per via ematica, di regola ai polmoni. Le metastasi allo scheletro sono rare e ancor più rare sono quelle linfonodali.
Figura 04

Figura 04: Quadro RM di osteosarcoma della tibia prossimale che mostra la disomogeneità strutturale del tumore e la distruzione della corticale metaepifisaria con infiltrazione dei tessuti molli. Sul versante opposto si rileva un’iniziale reazione periostale (←) e l’elevazione del periostio (⇽), che può portare alla formazione del cosiddetto “triangolo di Codman”.
Anatomia patologica
- Nella maggior parte dei casi l’osteosarcoma si presenta come un tumore voluminoso, a limiti mal definiti, che invade la cavità midollare e il rivestimento corticale, infiltrando i tessuti molli adiacenti. Il colore della massa neoplastica è bianco-grigiastro e al suo interno sono spesso presenti aree emorragiche, necrotiche o cistiche (Figura 03c). La consistenza è variabile: dura nelle zone osteogeniche, più molle in quelle dove predomina la componente fibroblastica, cartilaginea o mixoide.
- Il quadro microscopico può essere anch’esso molto vario in relazione al tipo cellulare più rappresentato. Circa il 50% degli osteosarcomi classici presenta aspetti predominanti di osteogenesi (forme osteoblastiche), mentre i rimanenti mostrano una prevalente differenziazione in senso condroide (forme condroblastiche) o una predominanza fibroblastica o fibro-istiocitica (forme fibroblastiche). Tali aspetti possono rendere l’osteosarcoma quasi indistinguibile dal condrosarcoma, dal fibrosarcoma o dall’istiocitoma fibroso maligno.
- L’aspetto istologico è in ogni caso caratterizzato dalla presenza di un parenchima mesenchimale anaplastico, con produzione di matrice osteoide e/o osso (in maggiore o minore misura) da parte delle cellule neoplastiche. Su questa caratteristica, che deve essere ricercata nell’osservazione microscopica di tutta la massa tumorale, si basa la diagnosi di osteosarcoma.
- La diagnosi differenziale deve prendere in considerazione altre neoplasie maligne dello scheletro come il sarcoma di Ewing, il condrosarcoma o alcune forme metastatiche.
Osteosarcomi meno frequenti
- L’osteosarcoma parostale origina dalla superficie dell’osso, con un’abbondante produzione di osso denso e un basso grado di anaplasia. È considerato un osteosarcoma a bassa malignità, pur potendo progredire verso una franca aggressività. Si localizza quasi esclusivamente alle metafisi delle ossa lunghe (nel 60% dei casi sul versante posteriore del femore distale); colpisce soprattutto soggetti di 20-50 anni di sesso femminile. Raramente insorge prima del termine della crescita.
- L’osteosarcoma periosteo è un osteosarcoma a bassa malignità e a predominanza condroblastica. Origina dal periostio delle ossa lunghe (femore, tibia) in sede diafisaria. Ha una crescita lenta, come la forma parostale, e si sviluppa in superficie senza invadere il canale midollare, aspetto ben evidente con la TC o la RM. L’osteosarcoma teleangectasico (o emorragico) rappresenta circa il 5% di tutti gli osteosarcomi È una forma totalmente osteolitica, caratterizzata da una struttura simil-spongiosa con ampie lacune emorragiche e una scarsa e immatura osteogenesi. Le aree necrotico-emorragiche possono essere così estese da rendere difficile il riscontro di cellule vitali per valutarne il grado di anaplasia. Le fratture patologiche sono frequenti e la prognosi è sovrapponibile a quella dell’osteosarcoma classico.
Terapia
- Il trattamento dell’osteosarcoma è multidisciplinare: è, infatti, necessaria una stretta collaborazione tra il chirurgo ortopedico e l’oncologo, poiché alla tradizionale terapia chirurgica deve essere associata la chemioterapia pre e postoperatoria.
- La terapia antiblastica ad alte dosi prima dell’intervento ha lo scopo di indurre la necrosi delle cellule tumorali, ridurre il volume della massa neoplastica e favorirne una migliore definizione dei margini. Questa condotta terapeutica ha consentito di ridurre il ricorso a interventi demolitivi (amputazioni), ottenendo nel contempo un significativo aumento delle percentuali di sopravvivenza.
- Inoltre, la prognosi funzionale di un arto sottoposto ad ampia resezione ossea è oggi migliorata dalla disponibilità di trapianti ossei massivi e protesi da ampia resezione.
Sarcoma DI ewing
- Il sarcoma di Ewing (SE) è la seconda neoplasia maligna per frequenza nei bambini e giovani adulti. La sua origine è rimasta sconosciuta per lungo tempo, ma oggi è classificato tra i tumori periferici primitivi neuroec-todermici (PNET), un gruppo di neoplasie che si ritiene derivi da elementi cellulari della cresta neurale.
- È tre volte meno frequente dell’osteosarcoma, con una netta predilezione per la seconda decade di vita: circa il 90% dei casi si manifesta in soggetti tra i 5 e i 25 anni.
- L’incidenza è lievemente maggiore nel sesso maschile, come gran parte dei tumori ossei primitivi.
Nessun distretto corporeo è immune dal SE, che si localizza in uguale misura nelle ossa piatte (bacino, scapola, coste), corte (vertebre) e lunghe (femore, tibia, omero ecc.). In queste ultime tende a svilupparsi in sede diafisaria, ma talvolta la crescita può essere così estesa da interessare l’intero segmento scheletrico. Più rara è la localizzazione al cranio, alle mani e ai piedi.
Quadro clinico
- Il SE si caratterizza, rispetto agli altri tumori scheletrici, per la presenza di una sintomatologia generale (febbre intermittente) con alterazione degli esami di laboratorio (leucocitosi, anemia, VES elevata) in circa la metà dei casi. Questi reperti si accompagnano a dolore e tumefazione locale, simulando un’osteomielite.
Diagnostica per immagini
- L’esame radiografico mostra piccole chiazze osteolitiche centrali, sfumate, che tendono a confluire conferendo all’osso un aspetto tarlato; la corticale viene quindi erosa, interrotta e superata dalla massa neoplastica (Figura 05). Solo di rado il tumore resta endomidollare per lungo tempo o mostra un’aumentata densità dell’osso.
- La reazione periostale nei pazienti più giovani è comune, ma le classiche immagini “a bulbo di cipolla” o “a dente di pettine” sono oggi osservate sempre più
- raramente, verosimilmente per la maggiore precocità della diagnosi.
Se non trattata, la neoplasia è a rapida evoluzione con metastasi precoci polmonari, scheletriche, linfonodali e cerebrali.
Figura 05

Figura 05: Sarcoma di Ewing del terzo distale della diafisi femorale.
Anatomia patologica
- L’aspetto macroscopico è quello di un tessuto grigiastro, encefaloide, con zone emorragiche e necrotiche. Queste ultime possono dare origine a materiale colliquato, che può simulare un processo suppurativo osteo-midollare.
- Istologicamente il tumore è costituito da un tappeto uniforme di piccole cellule rotonde, fittamente stipate, senza alcuna matrice intercellulare. Le cellule hanno un nucleo rotondo e voluminoso, mentre il citoplasma è scarso e mal definito.
- La positività alla colorazione PAS, soprattutto dopo fissazione in alcol, è caratteristica del SE ed è indicativa della presenza di abbondante glicogeno citoplasmatico. Tale reazione costituisce un elemento utile per la diagnosi differenziale con il linfoma maligno dell’osso e con le metastasi da neuroblastoma. Queste ultime si differenziano anche per la presenza di vere rosette (al contrario del SE dove si osserva una disposizione a pseudorosetta delle cellule intorno a capillari o elementi cellulari necrotici) e per l’elevazione dei cataboliti delle catecolamine (acido omovanilico e vanilmandelico) nelle urine.
- Alcune indagini immunoistochimiche e ultrastrutturali permettono oggi una maggiore caratterizzazione del SE.
Terapia
- Il SE è particolarmente sensibile alla radioterapia e alla chemioterapia, ma anche la chirurgia gioca un ruolo fondamentale nel trattamento di questa neoplasia. Per- tanto il trattamento si basa sulla combinazione di queste tre modalità terapeutiche: chemioterapia (sempre), chirurgia (quando praticabile) e radioterapia (in alternativa all’intervento o associato a esso in caso di resezione non radicale del tumore).
- Analogamente a quanto praticato per l’osteosarcoma, la chemioterapia è eseguita sia prima sia dopo la chirurgia, sia essa conservativa o demolitiva.
Istiocitoma fibroso maligno
- L’istiocitoma fibroso maligno (IFM) è un tumore maligno di origine istiocitaria, costituito da cellule di aspetto simil-fibroblastico e simil-istiocitario; può originare sia dallo scheletro sia dai tessuti molli (Figura 06) e in passato è stato accomunato al fibrosarcoma.
È relativamente frequente (5% di tutte le neoplasie maligne ossee primitive), interessando in maggiore misura l’età media e avanzata (80% dei casi nei soggetti di 40-70 anni) con una lieve predilezione per il sesso maschile. - La localizzazione preferenziale è rappresentata dalla metafisi delle ossa lunghe, soprattutto quelle in prossimità del ginocchio.
L’IFM può essere primitivo oppure secondario a lesioni preesistenti (anziani), in particolare a malattia di Paget, infarti ossei e terapia radiante.
Figura 06

Figura 06: Istiocitoma fibroso maligno dei tessuti molli (↗), infiltrante le strutture miofasciali mediali della coscia destra.
Quadro clinico
- La sintomatologia è aspecifica, con dolore e tumefazione a comparsa più o meno precoce. Le fratture patologiche sono frequenti (circa 3/4 dei casi nell’evoluzione della malattia).
Diagnostica per immagini
- Il quadro radiografico è quello di un’osteolisi pura, irregolare, a limiti sfumati, localizzata in sede centrale o periferica all’interno del canale midollare (Figura 07). La massa neoplastica provoca l’erosione e l’interruzione della corticale, mentre la reazione periostale è scarsa. L’area osteolitica tende a espandersi in tempi brevi verso la diafisi e l’epifisi. Le metastasi ossee e polmonari sono frequenti.
Figura 07

Figura 07: Istiocitoma fibroso maligno della metafisi prossimale della tibia: la corticale non appare ancora erosa e non si osserva alcuna reazione periostale.
Anatomia patologica
- L’IFM si presenta come una massa di colore biancastro, con zone giallastre o brune a causa di fenomeni necrotici ed emorragici, e di consistenza soffice, parenchimatosa.
- L’istologia mostra due popolazioni cellulari, fibroblastica e istiocitaria, espressione del medesimo clone neoplastico. Il contingente istiocitario è costituito da cellule grandi, globose, con abbondante citoplasma eosinofilo; il polimorfismo è frequente con cellule e
- Mitosi atipiche. Si possono anche osservare cellule giganti multinucleate di significato reattivo.
La componente fibroblastica è rappresentata da elementi fusati, disposti in maniera “storiforme”, con abbondante produzione di collageno. - Alla periferia è presente un infiltrato infiammatorio, costituito per lo più da linfociti.
Terapia
- Il trattamento si avvale, come per altri sarcomi, della chirurgia (resezione ampia) e della chemioterapia adiuvante pre- e postoperatoria. Con questi protocolli terapeutici le percentuali di sopravvivenza sono simili a quelle raggiunte nell’osteosarcoma (intorno al 60-70% dei casi).