Dettagli
- Inquadramento diagnostico
- MM asintomatico
- MM sintomatico
- Mieloma multiplo sintomatico: trattamento
- Criteri di risposta alla terapia
Il Mieloma Multiplo (MM)
Il Mieloma Multiplo (MM) è una neoplasia caratterizzata dalla proliferazione di un clone di plasmacellule neoplastiche. Le plasmacellule si accumulano a livello midollare e producono immunoglobuline tutte dello stesso tipo (componente monoclonale, CM). Le cause del MM non sono note e la sua patogenesi è sostenuta da complessi biologici ancora in via di definizione. Esistono sporadiche segnalazioni di MM familiare. La maggior parte dei casi esordisce come MM de novo, anche se sembra possibile, virtualmente postulare in tutti i pazienti, una precedente fase di Gammopatia monoclonale di incerto significato (MGUS) (1). In Italia il MM rappresenta 1,2% di tutti i tumori diagnosticati tra gli uomini e 1,3% tra le donne con un’incidenza media annuale di 9,5 casi ogni 100.000 uomini e 8,1 casi ogni 100.000 donne. Le stime indicano un totale di 2.315 nuovi casi diagnosticati ogni anno fra i maschi e di 2.098 casi fra le femmine. I tassi d’incidenza per MM sono abbastanza omogenei sia come distribuzione regionale sia come andamento nel tempo. L’incidenza del MM è nel complesso stabile mentre la mortalità è in lieve calo (figura B). Il MM è una patologia dell’età avanzata (figura A): infatti, l’età mediana alla diagnosi è di 68 anni, circa il 2% dei pazienti all’esordio ha meno di 40 anni mentre il 38% dei pazienti ha un’età superiore a 70 anni.
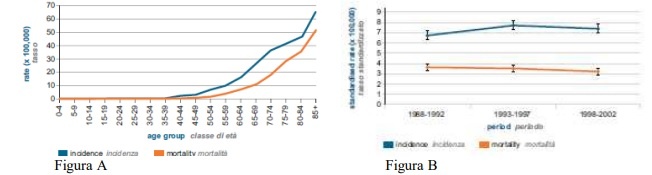
Secondo i dati epidemiologici dall’AIRTUM recentemente aggiornati, l’incidenza globale nel 2015 è rimasta stabile (8 casi per 100000 abitanti), la sopravvivenza globale a un anno è del 76%, mentre a cinque anni è solo del 42%. Non si registrano differenze sulla base del sesso o della distribuzione geografica (2-3).
Inquadramento diagnostico
Al riscontro di una CM sierica è indicato sottoporre il paziente ad esami di screening di I livello, un’accurata anamnesi e ad una valutazione clinica. La presenza di una CM può associarsi a MM così come ad altri quadri clinici patologici e non, quali:
• altri tumori di origine linfoide e mieloide (es. leucemia linfatica cronica/linfomi a basso grado, leucemia mieloide cronica, mielodisplasia);
• tumori solidi (es. carcinoma del colon e della mammella);
• malattie non neoplastiche (es. Cirrosi epatica, sarcoidosi, morbo di Gaucher, pioderma gangrenoso);
• malattie autoimmuni (es. artrite reumatoide, miastenia gravis, malattia da crioagglutinine);
• malattie infettive (es. tubercolosi, infezioni virali, parassitosi);
• altre discrasie plasmacellulari (es. MGUS, morbo di Waldenström, amiloidosi, POEMS Syndrome);
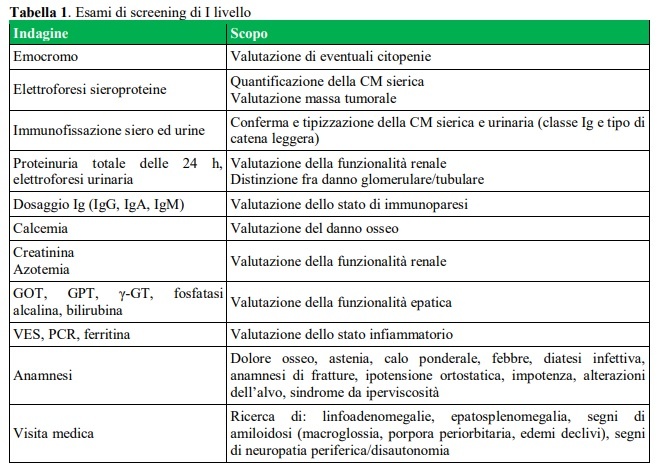
Gli approfondimenti di I livello comprendono l’esecuzione di esami di laboratorio, sierici ed urinari, come schematizzato in tabella 1.
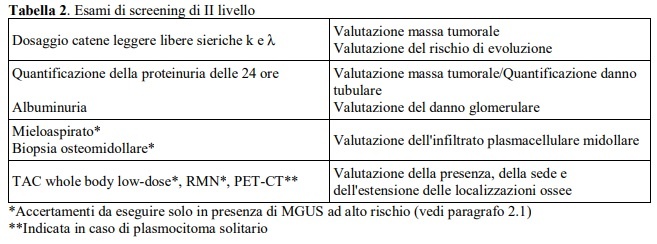
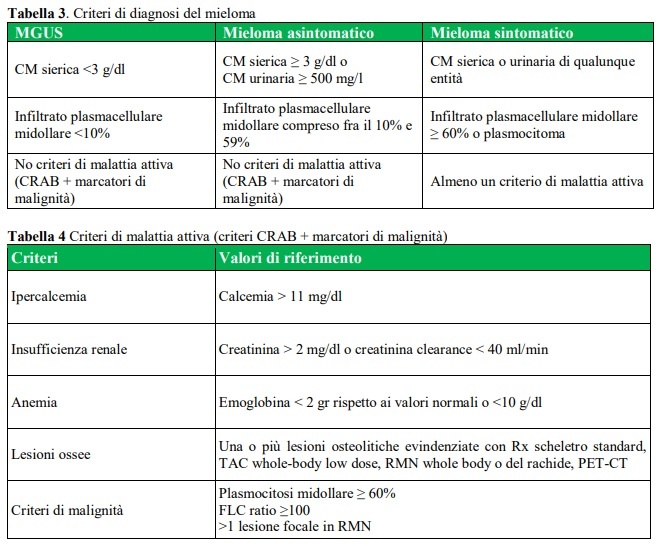
Nel caso di una CM di tipo IgM, frequentemente correlata a patologie linfoproliferative a basso grado (es. linfomi a basso grado, LLC e malattia di Waldenstrom), è utile integrare l’inquadramento diagnostico con esami ematici e radiologici ad hoc. Qualora la CM fosse tipizzata come IgG, IgA, o come sola catena leggera (k o λ) il paziente dovrà eseguire esami screening di II livello (tabella 2) che permettono una diagnosi differenziale, in accordo con l’International Myeloma Working Group, tra diverse situazioni cliniche: MGUS, Plasmocitoma solitario, MM asintomatico e MM sintomatico (tabella 3 e tabella 4) (4 – 8).
MM asintomatico
Il MM si definisce asintomatico secondo i seguenti criteri (Tabelle 3 e 4) (8) (Livello di evidenza 4):
• presenza di CM sierica ≥3 g/dl o CM urinaria ≥ 500 mg/24 h e/o plasmocitosi midollare compresa fra 10%-59% e
• assenza di criteri di “danno d’organo” ([C]alcium elevation, [R]enal insufficiency, [A]nemia, [B]one lytic lesion, CRAB) e dei, cosidetti, marcatori di malignità (plasmocitosi midollare ≥ 60%, FLC ratio ≥ 100, lesioni focali > 1 alla RMN).
La popolazione di pazienti affetti da MM asintomatico è molto eterogenea in quanto include pazienti a basso rischio evolutivo, che al pari delle MGUS non richiedono trattamento per molti anni, e pazienti che, al contrario, evolvono in mieloma sintomatico in un tempo breve. (Livello di evidenza 3). In questi pazienti viene consigliata una sorveglianza clinico-strumentale ravvicinata (4-5) (Livello di evidenza 3), per evidenziare precocemente un’eventuale evoluzione a malattia sintomatica (10 – 14).
Fra i fattori associati ad un significativo rischio di progressione a MM sintomatico (rischio di progressione ≥ 50% a 2 anni dalla diagnosi), vi sono: CM sierica ≥ 3 g/dl, IgA MM, l’immunoparesi, un pattern evolutivo di malatta (aumento della CM/catene leggere libere sieriche ≥25% a 2 controlli successivi a distanza di 6 mesi), alterazioni citogenetiche (t(4;14), del(17p) o 1q amplificazione), presenza di lesioni con “pattern diffuso” o 1 lesione focale alla RMN, lesioni captanti in PET-CT senza evidenza di lesioni osteolitiche sottostanti (15). Gli studi controllati che hanno valutato il ruolo del trattamento precoce nel paziente asintomatico hanno evidenziato un miglioramento del tempo alla progressione senza evidenziare un beneficio in termini di sopravvivenza globale (16-18). Un recente studio clinico di fase III condotto in pazienti con MM asintomatico ad alto rischio evolutivo (124 pazienti totali) ha valutato l’efficacia di una terapia con Lenalidomide + Desametasone versus nessun trattamento) (19). Dopo un follow-up mediano di 32 mesi, viene riportata una evoluzione a MM sintomatico in 9 pazienti (15%) nel braccio di trattamento contro in 37 pazienti (59%) nel braccio di osservazione (p <0,0001). La sopravvivenza globale stimata a 3 anni è stata del 93% nel braccio di trattamento e il 76% nel braccio di controllo (P = 0.04). E’ necessario attendere un periodo di osservazione più lungo per valutare il reale beneficio del trattamento a fronte di eventuali tossicità a lungo termine.
Concludendo, al difuori di studi clinici controllati, nei pazienti con MM asintomatico è attualmente indicato il solo monitoraggio clinico strumentale modulando la tempistica in base al rischio di evolutività del paziente, mentre non è indicato alcun trattamento chemioterapico nè con bisfosfonati, (7-8). (Livello di evidenza 4).

MM sintomatico
Nei pazienti con MM sintomatico, alle caratteristiche cliniche e laboratoristiche diagnostiche si associano uno o più segni di malattia attiva (8).
Nel paziente con MM sintomatico, andranno eseguite indagini di approfondimento (Tabella 5), che possono aiutare il clinico nel corretto inquadramento diagnostico e prognostico della patologia.
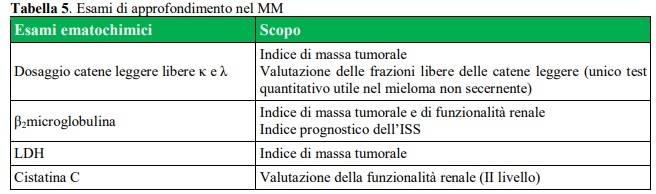
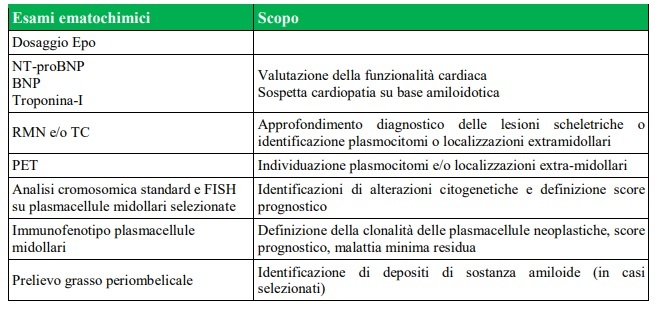
Per la definizione prognostica, infine, viene eseguita una stadiazione di malattia seguendo i criteri di Durie e Salmon (DS) (13) e dell’International Staging System (ISS) (14), quest’ultimi recentemente aggiornati (RISS) (20) (tabella 6).
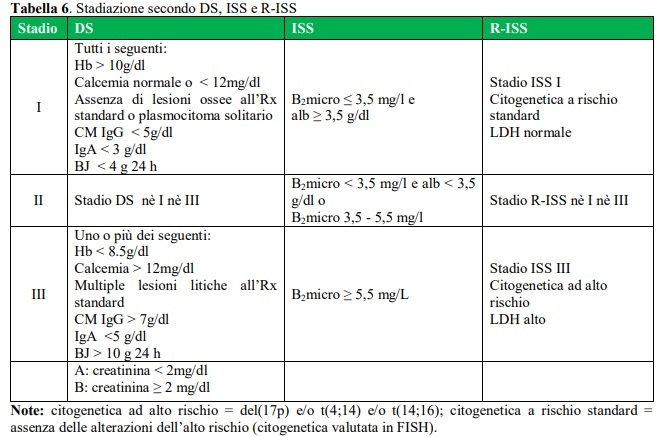
Altri fattori prognostici identificati come significativamente associati all’outcome sono la qualità della vita (20), l’età (21). La presenza di alterazioni cromosomiche alla citogenetica standard e all’immunofluorescenza (FISH) permette di identificare specifiche classi di rischio (22).
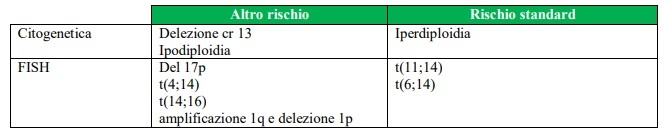
In pazienti con MM sono state evidenziate sia traslocazioni sia delezioni: la delezione del cromosoma 13, è stata riportata correlarsi con una breve durata sia della sopravvivenza libera da malattia che di quella globale (23). La delezione del cromosoma 17p13 (locus del tumor suppressor gene p53) caratterizza pazienti ad alto rischio (24). Sono riportate diverse alterazioni cromosomiche che coinvolgono il braccio lungo del cromosoma 14, (14q32, ove è presente il gene per le catena pesanti delle Immunoglobuline), con prognosi differenti (t(4;14) (p16;q32) – t(14;20)(q32;q11) – t(14;16)(q32; q23) – t(11;14)(q13; q32). Tra queste, la traslocazione t(4;14) si correla con la peggior prognosi, anche nel setting dei pazienti trattati con terapia ad alte dosi (25) (Tabella 7) (Livello di evidenza 3). Più recentemente sono state identificate alterazioni cromosomiche del cromosoma 1 (amplificazione 1q e delezione 1p) anch’esse prognosticamente negative. (26).
Il sintomo più frequente alla diagnosi di MM rimane in dolore osseo, correlato a danno d’organo (lesioni osteolitiche o a fratture/crolli vertebrali). Il paziente con MM sintomatico va avviato il prima possibile ad un trattamento sistemico (vedi Trattamento). In caso di situazioni cliniche particolari (fratture patologiche, Ipercalcemia e insufficienza renale acuta) vanno, inoltre, avviate le procedure d’urgenza del caso (vedi terapia di supporto).
Mieloma multiplo sintomatico: trattamento
Nel paziente affetto da MM sintomatico, cioè che presenta segni correlati a patologia attiva (CRAB/marcatori di malignità, Tabelle 3-4), il trattamento è sempre indicato. Gli scopi del trattamento sono l’ottenimento di risposte il più possibile profonde, il miglioramento della progressione libera da malattia (progression free survival, PFS) e la sopravvivenza globale (overall survival, OS) e, in pazienti frail, anche il solo miglioramento della qualità della vita ed il controllo della malattia. Tali obiettivi si possono ottenere mediante la combinazione di trattamenti efficaci con un’adeguata terapia di supporto.
L’Introduzione dei nuovi farmaci nel prontuario terapeutico per il MM, come Talidomide, Lenalidomide, Bortezomib e Pomalidomide, spesso in combinazione con Desametasone, ha portato ad un miglioramento della sopravvivenza, sia nei pazienti alla diagnosi sia alla recidiva della malattia (vedi paragrafi successivi).
I pazienti con MM sintomatico sono avviati a trattamento di prima linea diversificato in relazione all’età anagrafica, all’eventuale presenza di comorbità e al loro performance status. I pazienti di età ≤ 70 anni con normale funzionalità cardiaca, polmonare ed epatica e senza significativo rischio infettivo sono candidabili ad un trattamento più intensificato comprensivo di trapianto autologo di cellule staminali emopoietiche (ASCT). Va sottolineato come l’insufficienza renale, possibile complicanza del MM alla diagnosi, non costituisca una controindicazione assoluta a trattamenti intensificati.
Nel paziente candidato ad ASCT, farmaci tossici per il compartimento staminale, quali alchilanti e Lenalidomide (per un tempo di trattamento superiore ai sei mesi), non dovrebbero essere somministrati nella fase di induzione pre trapianto per non compromettere la mobilizzazione e la successiva raccolta di cellule staminali emopoietiche. Pertanto, di fronte ad una nuova diagnosi di MM sintomatico, è fondamentale una valutazione globale del paziente (età, patologie associate, performance status) allo scopo di definirne il programma di trattamento.
Nei pazienti di età > 70 anni, gli obiettivi del trattamento, precedentemente elencati, devono comprendere una opportuna valutazione della fragilità al fine di apportare modifiche al dosaggio dei farmaci prescritti per aumentare la tollerabilità del trattamento.
Criteri di risposta alla terapia
Una corretta valutazione della risposta al trattamento è essenziale nella gestione del MM. Attualmente vengono applicati i criteri proposti nel 2006 dall’ International Myeloma Working Group (IMWG) (tabella 8).


Abbreviazioni: sCR = risposta completa stringente; CR = risposta completa; PR = risposta parziale; VGPR = risposta parziale molto buona; SD = malattia stabile; PD = progressione di malattia; FLC = catene leggere libere.
Note:
1 Tutte le categorie di risposta richiedono due esami consecutivi fatti in qualsiasi momento prima dell’inizio di una nuova terapia; tutte le categorie richiedono inoltre l’assenza di nuove o progressive lesioni ossee (se erano stati eseguiti gli studi radiografici). Gli studi radiografici non sono richiesti per soddisfare i criteri di risposta.
2 Non è necessaria la ripetizione della biopsia per conferma.
3 La presenza/assenza di cellule clonali è basata sul rapporto kappa/lambda (è richiesta l’analisi all’immunoistochimica o all’immunofluorescenza di almeno 100 plasmacellule). Un rapporto kappa/lambda anormale è >4:1 e <1:2.
4 Aumenti della componente monoclonale > 1 g/dl sono sufficienti a definire la recidiva se la componente di partenza era > 5 g/dl.
5 La recidiva dalla CR ha il cut-off al 5% vs 10% delle altre categorie di risposta.
Fonte: Linee Guida AIOM – aiom.it