Dettagli
- Aspetti epidemiologici
- Genetica
- Patogenesi della malattia polmonare
- Manifestazioni cliniche
- Fibrosi cistica classica e non classica
- Diagnosi
- Prognosi
- Trattamento
Fibrosi cistica
- La fibrosi cistica (FC) è la più comune malattia genetica a prognosi infausta nella razza caucasica. Si trasmette con carattere autosomico recessivo ed è causata dalla mutazione di un gene che codifica per una proteina chiamata “Cystic fibrosis transmembrane conductance regulator” (CFTR), la cui principale funzione è quella di rappresentare un canale del cloro a livello della superficie apicale delle cellule epiteliali e di regolare gli scambi idroelettrolitici transmembrana. È una malattia sistemica con interessamento di numerosi organi e apparati ed eterogeneità fenotipica delle manifestazioni cliniche; la malattia polmonare è la principale causa di morbilità e mortalità legate alla malattia ed è caratterizzata principalmente da ostruzione cronica delle vie aeree, infezione bronchiale cronica e successivo sviluppo di bronchiectasie con progressivo deterioramento della funzione respiratoria fino all’insufficienza respiratoria.
Aspetti epidemiologici
- L’incidenza della FC tradizionalmente era stimata essere 1 su 2500-3000 nati vivi nella popolazione di origine europea; tuttavia, oggi, i dati dei programmi di screening neonatale rivelano un’incidenza più bassa che in passato e stimata in media tra 1/3000-1/6000 in tali popolazioni, con una frequenza di portatori riportata tra 1/28 e 1/40. L’incidenza della malattia varia tuttavia nelle varie aree geografiche e nelle diverse etnie; essa è rara in alcune popolazioni di origine asiatica con un’incidenza riportata di 1/350000 in Giappone.
- L’epidemiologia della malattia è notevolmente mutata negli ultimi decenni: l’aumento delle conoscenze legato ai notevoli progressi in campo genetico e alle recenti acquisizioni di biologia cellulare e molecolare e le migliorate strategie di trattamento hanno portato da un lato a un drammatico aumento della sopravvivenza dei pazienti affetti dalla malattia dall’altro alla diagnosi di forme non classiche con manifestazioni cliniche più lievi che talora arrivano misconosciute in età adulta e che si associano a una sopravvivenza maggiore. Ne è derivato un significativo aumento della popolazione di pazienti adulti affetti dalla malattia e tale fenomeno aumenterà ulteriormente negli anni futuri.
Genetica
- Il gene della FC, denominato Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) è localizzato a livello del braccio lungo del cromosoma 7 ed è costituito da 27 esoni. Esso codifica la sintesi di una proteina di 1480 residui aminoacidici, denominata anch’essa CFTR, la cui funzione principale, come già accennato, è quella di rappresentare un canale transepiteliale per il cloro (Immagine 01).
- Sono state a oggi identificate più di 2200 mutazioni del gene CFTR e il loro numero tende continuamente a crescere, sebbene solo per un piccolo numero di esse sia nota l’importanza funzionale. La mutazione più frequente è la mutazione DF508 che causa la delezione del residuo fenilalaninico in posizione 508 ed è responsabile di circa due/terzi degli alleli mutati nelle popolazioni del Nord Europa e del Nord America. È tuttavia da sottolineare che la frequenza relativa di questa e delle altre mutazioni varia considerevolmente tra differenti gruppi etnici e aree geografiche. Le altre mutazioni conosciute sono molto meno frequenti e a volte legate all’appartenenza a un particolare gruppo etnico.
- In base ai meccanismi biomolecolari attraverso i quali le mutazioni del gene causano il deficit quantitativo o funzionale della proteina, esse sono state raggruppate in sei classi principali, che si associano a una diversa severità di malattia:
- mutazioni di classe I in cui si verifica un difetto di sintesi della proteina;
- mutazioni di classe II che si associano a un difetto di processazione e maturazione della proteina;
- mutazioni di classe III caratterizzate da un difetto di regolazione;
- mutazioni di classe IV che comportano un difetto di conduttanza;
- mutazioni di classe V che determinano un parziale difetto nella produzione della proteina o nel suo trasporto a livello di membrana;
- mutazioni di classe VI, con produzione di una proteina funzionale ma instabile, che va incontro ad accellerata degradazione.
- Le mutazioni di classe I, II, III sono in genere associate a un fenotipo più severo di malattia e a insufficienza pancreatica, mentre quelle di classe IV, V e VI, che comportano una qualche funzione residua della proteina CFTR, si associano a fenotipi clinici più lievi, con sufficienza pancreatica.
- Gli studi più recenti hanno messo in evidenza come la relazione tra genotipo e fenotipo clinico è stretta per la malattia pancreatica, ma è più lieve per la malattia polmonare e hanno suggerito che, accanto al difetto del gene CFTR, alterazioni di geni diversi dal CFTR (geni modificatori) e fattori ambientali contribuiscano a spiegare l’eterogeneità fenotipica della malattia.
- La proteina CFTR è espressa, in modo non uniforme, a livello di diversi epiteli, quali quello delle vie aeree, del tratto gastrointestinale, dell’apparato riproduttivo, del dotto sudoriparo.
- Sebbene la sua principale funzione sia quella di rappresentare un canale per il cloro, regolato da cAMP e posto sulla membrana apicale delle cellule epiteliali, studi recenti hanno evidenziato come tale proteina abbia molteplici altre funzioni, attraverso cui interviene nella regolazione del trasporto ionico transmembrana e in molteplici altri processi cellulari. In particolare, è stata dimostrata un’azione inibitoria della CFTR sul canale per il sodio ENaC (Endothelial Natrium Channel) e un effetto modulatorio su altri canali del cloro. Essa è inoltre coinvolta nel trasporto di bicarbonato così come di altre proteine quali il glutatione e interagisce con altre proteine intracellulari. Studi in vitro e in vivo suggeriscono inoltre che la proteina è implicata, direttamente o indirettamente, anche nei complessi meccanismi che sono alla base dell’aumentata suscettibilità all’infezione bronchiale batterica e della esagerata e persistente risposta infiammatoria che caratterizza la malattia.
Immagine 01
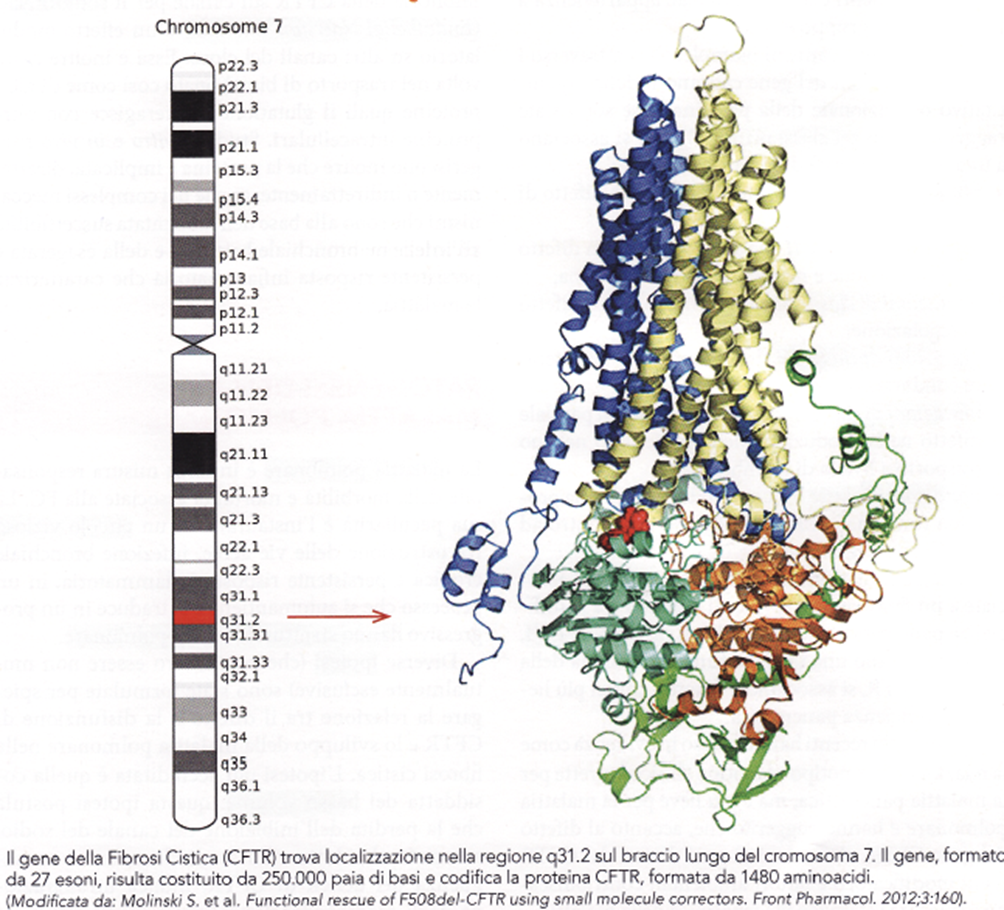
Immagine 01. Gene CFTR.
Patogenesi della malattia polmonare
- La malattia polmonare è in larga misura responsabile della morbilità e mortalità associate alla FC. La sua peculiarità è l’instaurarsi di un circolo vizioso tra ostruzione delle vie aeree, infezione bronchiale cronica e persistente risposta infiammatoria, in un processo che si automantiene e si traduce in un progressivo danno strutturale broncopolmonare.
- Diverse ipotesi (che potrebbero essere non mutualmente esclusive) sono state formulate per spiegare la relazione tra il difetto o la disfunzione di CFTR e lo sviluppo della malattia polmonare nella fibrosi cistica. L’ipotesi più accreditata è quella cosiddetta del basso volume: questa ipotesi postula che la perdita dell’inibizione del canale del sodio, associata al ridotto efflusso di cloro, come conseguenza del deficit di CFTR, induca un’alterazione degli scambi idroelettrolitici a livello del lume bronchiale, con aumentato riassorbimento di sodio e acqua attraverso la superficie apicale delle cellule epiteliali cui consegue un’alterata idratazione del fluido di rivestimento delle vie aeree; la riduzione di volume del fluido periciliare indurrebbe a sua volta una compromissione della clearance mucociliare e aumento della densità e viscosità delle secrezioni bronchiali, favorendo pertanto l’infezione batterica.
- Studi in vitro hanno anche suggerito che il difetto o l’alterazione funzionale della proteina CFTR comprometta i meccanismi di difesa locali a livello delle vie aeree e contribuisca direttamente a facilitare l’infezione cronica bronchiale da parte di patogeni specifici. È stato infatti evidenziato che in condizioni fisiologiche la proteina CFTR ha un ruolo come recettore di superficie per l’internalizzazione di P. aeruginosa con conseguente eliminazione del microrganismo dalle vie aeree; l’eliminazione di P. aeruginosa attraverso tale meccanismo risulterebbe pertanto compromessa in condizioni di difetto della proteina. È stato inoltre documentato che il deficit ai CFTR induce un’aumentata espressione di recettori che legano P. aeruginosa e S. aureus, favorendo l’aumentata adesione batterica alle cellule epiteliali.
- Osservazioni recenti sia in vitro che in vivo hanno anche documentato che l’alterazione della proteina CFTR indurrebbe di per sé anche una disregolazione della risposta infiammatoria nelle vie aeree dei pazienti con fibrosi cistica, suggerendo che l’infiammazione bronchiale possa anche precedere l’infezione e svilupparsi molto precocemente nel corso della malattia.
- Indipendentemente dalla relazione temporale tra infiammazione e infezione, la marcata e persistente risposta infiammatoria presente nelle vie aeree dei pazienti con fibrosi cistica viene comunque esaltata dalla comparsa dell’infezione ed è in ultima analisi responsabile del progressivo danno delle strutture broncopolmonari. Le cellule infiammatorie e in particolare i neutrofili rilasciano infatti una serie di mediatori potenzialmente lesivi, in particolare enzimi proteolitici e radicali ossidanti, che contribuiscono in modo determinante al danno strutturale broncopolmonare.
Manifestazioni cliniche
- Come già accennato, la fibrosi cistica può manifestarsi con un’ampia variabilità fenotipica, con quadri clinici che differiscono in funzione degli organi interessati e della gravità delle lesioni indotte. Anche la sintomatologia di esordio, così come l’età di insorgenza dei sintomi, può variare notevolmente.
APPARATO RESPIRATORIO
- Le manifestazioni cliniche a carico dell’apparato respiratorio compaiono nella maggioranza dei casi entro il primo anno di vita; tuttavia l’età di insorgenza dei primi sintomi polmonari è variabile, potendo essi rendersi manifesti più tardivamente nell’età adulta.
- La sintomatologia di esordio è in genere aspecifica e comprende tosse inizialmente secca, quindi produttiva e infezioni respiratorie ricorrenti.
- Talora il sintomo di esordio può essere rappresentato da una complicanza della malattia quale l’emoftoe o il pneumotorace. I pazienti in cui la diagnosi viene posta in età adulta riferiscono spesso una lunga storia di disturbi respiratori persistenti, erroneamente diagnosticati e trattati come asma, bronchite cronica o bronchiectasie da causa non definita.
- Come già accennato, la caratteristica peculiare della malattia a livello broncopolmonare è costituita dall’infezione bronchiale cronica, spesso polimicrobica. Le infezioni respiratorie svolgono un ruolo cruciale nell’evoluzione della malattia. All’infezione cronica si sovrappongono ricorrenti episodi di esacerbazioni acute, che si manifestano principalmente con aumento della tosse e della produzione di escreato, eventualmente con dispnea o ridotta tolleranza allo sforzo fisico, anoressia e perdita di peso, riduzione degli indici di funzionalità respiratoria e talora si accompagnano a rialzo termico, in genere non marcato. Con il progredire della malattia, la frequenza delle esacerbazioni aumenta con progressivo declino della funzione respiratoria fino allo sviluppo dell’insufficienza respiratoria.
- Nella prima infanzia prevalgono le infezioni da S. aureus ed H. influenzae. Precocemente nel decorso della malattia, tuttavia, l’infezione predominante diventa quella da P. aeruginosa che complica il quadro clinico, inducendo la progressione del danno broncopolmonare. L’infezione, inizialmente intermittente, diviene successivamente cronica con la presenza di differenti tipi di colonie in genere con differente sensibilità agli antibiotici; con il tempo compaiono colonie mucoidi, tipiche della fibrosi cistica (i microrganismi crescono in colonie avvolte da un biofilm di alginato), difficili se non impossibili da eradicare con i trattamenti antibiotici standard. L’infezione bronchiale cronica amplifica il processo infiammatorio già presente nelle vie aeree, attraverso il rilascio di citochine e chemochine e l’ulteriore reclutamento di neutrofili. La prevalenza dell’infezione da P. aeruginosa aumenta con l’età; studi recenti hanno mostrato, tuttavia, che il microrganismo può essere isolato dal lavaggio broncoalveolare in una significativa percentuale di pazienti già entro i tre anni di età, anche in assenza di sintomi respiratori, suggerendo che il microrganismo può essere presente nelle vie aeree già prima che l’infezione cronica diventi manifesta.
- Negli ultimi decenni stanno assumendo un ruolo sempre più rilevante altri microrganismi quali B. cepacia (un complesso di almeno nove differenti specie), S. maltophilia, A. xylosoxidans, S. aureus meticillino-resistente, la cui principale caratteristica è la resistenza alla maggior parte degli antibiotici. Aspergillus fumigatus può essere isolato nell’escreato in corso di fibrosi cistica (fino al 50% dei pazienti) e fino al 10-15% dei pazienti presenta come complicanza della malattia l’Aspergillosi Broncopolmonare Allergica. Un altro gruppo di microrganismi che si isolano con maggior prevalenza nei pazienti adulti con FC sono i Micobatteri non tubercolari, in particolare Mycobacterium abscessus e Mycobacterium avium-intracellulare che in alcuni pazienti si associano a manifestazioni patologiche.
- Con il tempo, l’infezione cronica endobronchiale e le ripetute esacerbazioni determinano importanti alterazioni strutturali delle pareti bronchiali, con formazione di bronchiectasie, alterazioni tipiche della malattia, in genere prevalenti ai lobi superiori; si associano alterazioni di tipo enfisematoso e fibrosi, con conseguente progressivo deterioramento della funzione respiratoria fino all’insufficienza respiratoria (Immagine 02).
- Il decorso della malattia può essere complicato da episodi recidivanti di pneumotorace e da episodi di emoftoe in genere di modesta entità e in relazione alle riacutizzazioni infettive, ma talora anche massivi.
- Un’altra complicanza tipica della malattia e osservabile con particolare frequenza nell’età adulta è l’Aspergillosi Broncopolmonare Allergica, mentre il verificarsi di atelettasie lobari o segmentali è una complicanza prevalente nei primi anni di vita. Il clubbing è presente nella quasi totalità dei pazienti e in genere precocemente nel corso della malattia. Come già detto nelle fasi avanzate della malattia è presente l’insufficienza respiratoria e il cuore polmonare cronico.
- La radiografia del torace può evidenziare vari tipi di alterazioni come segni di iperinflazione polmonare, ispessimenti e dilatazione delle pareti bronchiali, addensamenti peribronchiali, impatti mucoidi, formazioni cistiche; essa non è tuttavia considerata una metodica sensibile per valutare le alterazioni della malattia. La tomografia computerizzata ad alta risoluzione (HRCT) è in grado di evidenziare anomalie strutturali precoci e di documentare in maniera molto più accurata e sensibile l’estensione e la severità delle alterazioni bronchiali e parenchimali.
- I test di funzionalità respiratoria sono indispensabili per definire l’entità della compromissione della funzione respiratoria e monitorare l’evoluzione della malattia. Mentre in fase precoce possono anche risultare nella norma, con il progredire del danno polmonare documentano inizialmente i segni dell'”air trapping”, quindi un quadro disventilatorio ostruttivo ingravescente, caratterizzato dalla riduzione del FEV1, del rapporto FEV1/FVC, dall’aumento del volume residuo (RV) e del rapporto tra RV e capacità polmonare totale (TLC) e dall’aumento delle resistenze al flusso aereo. Nelle fasi avanzate, la compromissione degli scambi gassosi comporta la comparsa di ipossiemia e, successivamente, di ipercapnia.
- Polipi nasali si riscontrano nel 15-20% dei pazienti. Quasi costante risulta il riscontro radiologico di opacamento dei seni paranasali, che è in genere clinicamente asintomatico.
Immagine 02
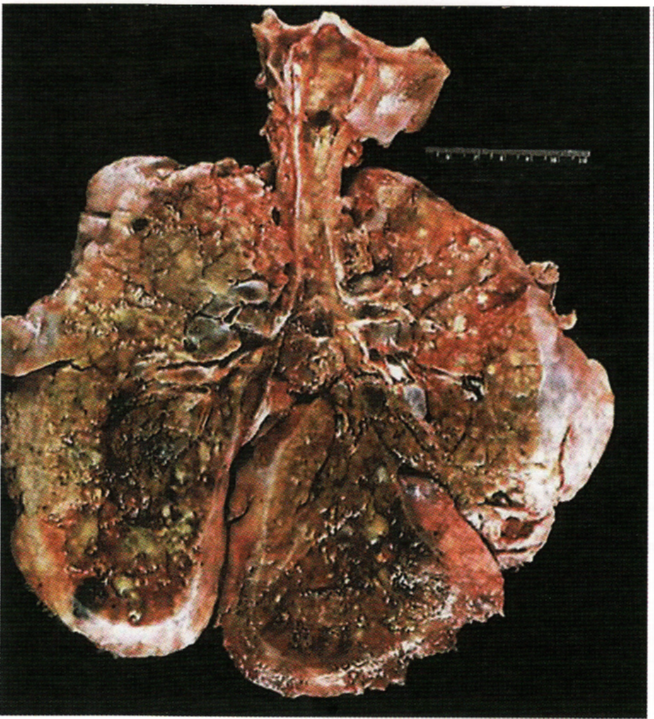
Immagine 02. Aspetto anatomo-patologico di polmone con fibrosi cistica.
APPARATO DIGERENTE
- L’ileo da meconio è la manifestazione più precoce di FC; si osserva in circa il 15% dei neonati affetti e può essere considerato una manifestazione patognomonica. Anche nelle età successive i disturbi della canalizzazione intestinale non sono rari; nel 15-20% dei pazienti si verificano episodi di subocclusione a livello dell’ileo terminale e del cieco, definiti sindrome da ostruzione dell’intestino distale (DIOS).
- In >90% dei pazienti affetti da FC è presente insufficienza pancreatica esocrina, che può essere già presente alla nascita o manifestarsi precocemente; nei pazienti con mutazioni di classe IV, V, o VI la funzione pancreatica rimane spesso conservata, sebbene alcuni sviluppino insufficienza pancreatica più tardivamente nel corso della vita. L’insufficienza pancreatica esocrina comporta maldigestione e malassorbimento soprattutto dei grassi, ma anche delle proteine; ne consegue una riduzione dell’apporto calorico con scarso o assente aumento di peso, ritardo di recupero del peso alla nascita nel neonato e deficit di accrescimento staturo-ponderale nelle età successive, meteorismo e distensione addominale, steatorrea. Si associa inoltre carenza di vitamine liposolubili.
- Episodi di pancreatite si verificano nel 15% circa dei pazienti con FC e funzione pancreatica esocrina conservata ed episodi recidivanti di pancreatite acuta possono talora condurre alla insufficienza pancreatica.
- Il 50% dei pazienti con FC presenta alterazioni epatiche; spesso si tratta di epatomegalia asintomatica, sostenuta da un quadro anatomopatologico di steatosi, ma una quota di questi pazienti presenta fibrosi biliare focale, che può progredire in cirrosi biliare in meno del 10% dei casi.
- L’aumento di viscosità della bile, secondario al difetto del trasporto di elettroliti e fluidi, alla presenza di mucine e alle alterazioni del metabolismo dei sali biliari, può provocare nel neonato ittero ostruttivo e nell’adulto diversi quadri anatomopatologici, tra cui la calcolosi colecistica.
SISTEMA ENDOCRINO
- Con l’aumentare dell’età, le alterazioni del pancreas esocrino possono progredire evolvendo verso la fibrosi, con interessamento delle insule la cui funzione viene compromessa. La ridotta produzione di insulina e glucagone induce una alterazione del metabolismo glucidico; inizialmente si osserva una ridotta tolleranza glucidica, quindi compare diabete franco; esso è denominato diabete FC correlato, in quanto non rientra né nel tipo I né nel tipo II. La sua prevalenza aumenta con l’età ed è stato stimato che fino al 40% dei pazienti adulti ne è affetto.
APPARATO RIPRODUTTIVO
- Nel maschio si osserva in circa il 98% dei casi sterilità per atresia dei deferenti, con azoospermia e ridotto volume dell’eiaculato. Nella femmina, nella maggior parte dei casi la fertilità è conservata.
APPARATO CARDIOCIRCOLATORIO
- Le alterazioni strutturali del parenchima polmonare e la conseguente insufficienza respiratoria condizionano un’ipertensione polmonare ingravescente con conseguente ipertrofia del ventricolo destro, episodi di scompenso cardiaco destro e cuore polmonare cronico.
APPARATO OSTEOARTICOLARE
- Diversi studi hanno dimostrato che circa il 30% dei pazienti affetti da FC presenta una riduzione della massa ossea, che oltre certi limiti determina un aumento del rischio di fratture anche per traumi di modesta entità. L’eziologia è multifattoriale: oltre alla disfunzione di CFTR, la malnutrizione, la deficienza di vitamina D, l’inattività fisica, l’infiammazione cronica, l’eventuale impiego della terapia steroidea, il diabete possono compromettere la deposizione ossea e favorire un incrementato riassorbimento osseo. Particolare attenzione deve essere quindi rivolta a favorire un adeguato apporto dietetico di calcio e di vitamina D, a favorire l’attività fisica e a monitorare adeguatamente i pazienti per la comparsa di tale complicanza.
- L’osteoartropatia ipertrofica si verifica in circa il 15 % dei pazienti in età adolescenziale e adulta.
Fibrosi cistica classica e non classica
- Come già sottolineato, la FC può manifestarsi con una ampia variabilità di manifestazioni fenotipiche in funzione degli organi interessati, della severità della malattia e dell’età di esordio della sintomatologia. Nella sua forma classica, la malattia è caratterizzata dall’associazione di broncopneumopatia cronica ostruttiva, insufficienza pancreatica esocrina, elevate concentrazioni di cloro e sodio nel sudore, familiarità per la malattia, infertilità maschile. Negli ultimi decenni, i progressi nell’ambito della genetica e della biologia molecolare e quindi nella conoscenza della malattia hanno portato all’identificazione di forme ”atipiche” o non classiche di FC che rappresentano una minoranza dei casi e in cui la diagnosi è più complessa. Queste forme includono quelle con insufficienza pancreatica esocrina e/o test del sudore normale o borderline, con manifestazioni cliniche che in genere diventano evidenti nell’età adolescenziale o adulta e con compromissione polmonare più lieve; queste forme si associano in genere a due mutazioni CFTR, almeno una delle quali risulta in funzione residua della proteina.
- Con sempre maggior frequenza vengono, infine, identificate condizioni patologiche associate a disfunzione di CFTR, ma in cui i criteri diagnostici necessari per la diagnosi di FC non sono soddisfatti.
- Queste condizioni, sono state definite come patologie CFTR correlate e sono verosimilmente il risultato dell’interazione di fattori ambientali e fattori genetici multifattoriali.
Diagnosi
- La Cystic Fibrosis Foundation Americana e il Working Group Europeo sulla diagnosi di fibrosi cistica hanno definito gli algoritmi diagnostici per la fibrosi cistica classica e non classica. Sulla base di tali linee guida, la diagnosi di FC si fonda sull’associazione di un quadro clinico caratteristico e dell’alterazione di tests di laboratorio che evidenziano un difetto della funzione CFTR, quali la positività del test del sudore, il riscontro di mutazioni note per causare la malattia su entrambi gli alleli e/o l’alterazione del test di differenza di potenziale nasale.
- La diagnosi è agevole nelle forme classiche con manifestazioni respiratorie, insufficienza pancreatica e test del sudore positivo. Al contrario, le forme non classiche, che sono sempre più numerose, risultano spesso di difficile inquadramento.
- A tutt’oggi, il test del sudore rimane il “gold standard” per la conferma diagnostica di FC; esso misura le elevate concentrazioni di cloro e di sodio nel sudore. È importante che il test sia eseguito correttamente, seguendo ‘accuratamente le linee guida, da personale esperto e in un laboratorio accreditato. Una concentrazione di cloro >60 mmol/l indica la positività del test, un valore <29 mmol/l è da considerarsi negativo, mentre il test è borderline per valori tra 30 e 59 mmol/l. I risultati devono essere interpretati anche in base all’età del soggetto, ma tipicamente una concentrazione di cloro >70 mmol/l negli adulti è in grado di discriminare tra fibrosi cistica e altre patologie polmonari. Il test deve essere ripetuto in caso di esito positivo o borderline. È da tener presente la possibilità di condizioni che ne inducono una falsa positività o negatività. Come già accennato, la negatività, anche ripetuta, del test del sudore non esclude tuttavia la diagnosi di FC: è infatti riconosciuto che esistono forme di FC con test del sudore borderline o costantemente negativo.
- La presenza di una mutazione del gene CFTR, nota per causare malattia, su entrambi gli alleli indica che un soggetto è affetto da FC; tuttavia, sebbene l’analisi genetica sia molto specifica per la diagnosi, essa non è del tutto sensibile a causa dell’alto numero di mutazioni. L’indagine genetica eseguita di routine è in grado di identificare le mutazioni più frequenti sul territorio nazionale e con l’inclusione delle mutazioni più frequenti ha una sensibilità di più del 90%; tuttavia con tale tipo di test non verranno diagnosticate le forme di malattia causate da mutazioni non screenate. D’altra parte, spesso nei soggetti con un quadro clinico compatibile con FC, ma test del sudore negativo o borderline, si riscontra all’indagine genetica una sola o nessuna mutazione genetica; infatti il test del sudore risulta spesso dubbio o negativo proprio in presenza di mutazioni rare. Oggi è possibile eseguire una analisi estensiva delle mutazioni dell’intero gene; si tratta però di una indagine costosa, che richiede tempi lunghi e talora propone risultati di difficile interpretazione; essa dovrebbe pertanto essere riservata ai soggetti con caratteristiche cliniche tipiche della malattia e test del sudore borderline e più in generale alle forme non classiche in cui vi sia un fondato sospetto clinico.
- Il test della differenza di potenziale nasale si basa sul principio che il difetto di secrezione di cloro e l’aumentato riassorbimento di sodio a livello della mucosa delle vie aeree comporta una alterata differenza di potenziale transepiteliale nei pazienti con FC. Il potenziale elettrico transepiteliale può essere misurato in vivo a livello del turbinato nasale inferiore. Sebbene tale metodica risulti molto utile nella diagnosi di FC, essa deve essere tuttavia ben standardizzata e non è disponibile in tutti i Centri.
SCREENING NEONATALE
- L’intento dello screening neonatale di massa è quello di attuare una diagnosi precoce della malattia, prima dell’insorgenza di complicanze nutrizionali importanti e soprattutto di danni polmonari irreversibili.
- Il test consiste nella determinazione, con metodo immunoenzimatico, dei livelli sierici di tripsinogeno, che risultano elevati nei neonati affetti da FC. Un test positivo è suggestivo, ma non diagnostico di FC; in questi casi per la conferma diagnostica è necessario effettuare ulteriori valutazioni, ripetendo il test dopo 1-3 settimane, effettuando il test del sudore e la ricerca di mutazioni del gene CFTR (che tuttavia si associa a problematiche di interpretazione nel caso si evidenzino mutazioni di significato clinico incerto).
Prognosi
- Come già accennato, negli ultimi decenni la sopravvivenza dei pazienti con FC è aumentata in modo significativo. L’aspettativa di vita, secondo i dati della Cystic Fibrosis Foundation era aumentata da 31 a 37 anni nella scorsa decade; studi recenti stimano che i nati con FC attualmente abbiano una aspettativa di vita di 50 anni o più.
- Le ragioni dell’aumentata sopravvivenza sono molteplici: sicuramente l’introduzione di nuovi antibiotici, le migliorate tecniche di fisioterapia respiratoria, il migliorato apporto nutrizionale e più recentemente l’avvento dei nuovi farmaci rivolti alla correzione del difetto di base della malattia e soprattutto il trattamento intensivo e globale del paziente nei Centri di riferimento specializzati per la diagnosi e la cura della malattia hanno avuto un ruolo determinante nel condizionare il miglioramento della prognosi della FC. Inoltre, le migliorate conoscenze sulla malattia e l’introduzione dello screening neonatale in molti paesi hanno comportato una riduzione dell’età a cui viene posta la diagnosi e conseguentemente un più precoce inizio del trattamento. Le migliorate conoscenze sulla malattia e lo sviluppo di nuovi metodi diagnostici permettono oggi, inoltre, di porre diagnosi di FC anche in forme non classiche, paucisintomatiche, per cui accanto a un reale miglioramento della prognosi, l’aumento della sopravvivenza è dovuto anche al riconoscimento di forme più lievi di malattia.
- Di fatto esiste una grande variabilità nell’evoluzione della malattia e quindi è difficile stabilire la prognosi nel singolo paziente. Questa dipende da fattori genetici, dallo stato nutrizionale, dalla compromissione della funzione respiratoria, dalla durata della infezione cronica da P. aeruginosa e dalla eventuale infezione da altri microrganismi difficili, dalle modalità di trattamento e dal sopraggiungere di complicanze.
Trattamento
- Dal momento della diagnosi il paziente con FC deve essere preso in carico presso un Centro specializzato, dove opera un team di personale medico-infermieristico dedicato e specializzato nella malattia e dove il paziente viene inserito in un programma di gestione complesso e integrato, secondo ben definite linee guida. Questo programma si prefigge lo scopo di controllare la malattia nelle sue differenti manifestazioni, con particolare attenzione alla rimozione delle secrezioni bronchiali, alla prevenzione e al trattamento dell’infezione bronchiale e delle manifestazioni infiammatorie a essa correlate, al controllo dello stato nutrizionale, alla prevenzione, diagnosi e trattamento delle complicanze.
TRATTAMENTO DELLE MANIFESTAZIONI RESPIRATORIE
- L’aumentata sopravvivenza e la ridotta morbilità della malattia osservate negli ultimi decenni sono in gran parte attribuibili all’impiego della terapia antibiotica mirata e aggressiva per la prevenzione e il trattamento dell’infezione polmonare.
- Alla luce dell’impossibilità di eradicare l’infezione cronica, una volta instauratasi, grande attenzione deve essere posta alla prevenzione della sua acquisizione con misure igieniche e di segregazione dei pazienti in coorti, in base alle risultanze microbiologiche dell’escreato, in modo da ridurre il rischio di infezioni crociate tra pazienti. Particolare attenzione, inoltre, sempre nell’ottica di prevenire la cronicizzazione dell’infezione, deve essere posta al trattamento precoce e aggressivo della prima colonizzazione da P. aeruginosa con l’impiego di antibiotici per via inalatoria (tobramicina o colistina) da soli o associati a chinolonici per via orale.
- La terapia antibiotica, in corso di riacutizzazione infettiva polmonare, è volta al ripristino delle condizioni clinico-funzionali di base e alla riduzione del processo infiammatorio e deve essere effettuata per via endovenosa, per almeno 14 giorni e sempre sulla base delle risultanze dell’esame batteriologico. In genere, il trattamento ottimale è costituito dall’impiego di associazioni di antibiotici con diverso meccanismo di azione per ridurre l’emergenza di ceppi resistenti. Nelle esacerbazioni più lievi può essere sufficiente l’impiego di antibiotici per via orale o inalatoria. In corso di riacutizzazioni è inoltre importante intensificare la fisioterapia respiratoria e porre particolare attenzione all’ottimizzazione dello stato nutrizionale.
- Ampiamente diffusa nella FC è la somministrazione di antibiotici per via aerosolica per il controllo dell’infezione cronica oltre che per il trattamento precoce dell’infezione da P. aeruginosa, con l’indubbio vantaggio di ridurre gli effetti sistemici e raggiungere elevate concentrazioni locali del farmaco.
- A dispetto della scarsità di studi controllati, la fisioterapia respiratoria riveste un ruolo di primo piano nella terapia della FC in quanto, rimuovendo le secrezioni bronchiali, ottiene sia una riduzione dell’ostruzione bronchiale, sia una azione di prevenzione del danno broncopolmonare mediante la riduzione della carica batterica e proteolitica endobronchiale. Diverse sono le tecniche impiegate; non esistono evidenze della superiorità di una tecnica rispetto a un’altra, per cui la scelta del tipo di fisioterapia è strettamente individuale e avviene in base alle caratteristiche cliniche nonché alle esigenze e preferenze del paziente, al fine di ottenere la maggiore efficacia e soprattutto la maggior compliance.
- L’impiego di desossiribonucleasi (DNAsi) ricombinante umana per via aerosolica quale mucolitico nella FC è basato sul fatto che l’elevata concentrazione di DNA derivante dalla lisi batterica e delle cellule infiammatorie contribuisce in maniera determinante alla aumentata viscoleasticità delle secrezioni bronchiali; con l’impiego cronico di tale farmaco si è osservato un certo grado di recupero funzionale e una significativa riduzione dell’incidenza di riacutizzazioni, pur con una ampia variabilità individuale.
- L’impiego di soluzione salina ipertonica che, agendo come agente iperosmolare, induce una reidratazione del fluido periciliare e quindi il miglioramento della clearance mucociliare si è dimostrato in grado di migliorare la funzione polmonare e ridurre le riesacerbazioni.
- Sulla base delle numerose evidenze di una intensa e persistente reazione flogistica a prevalente componente neutrofila a livello delle vie aeree dei pazienti con FC, che è presente molto precocemente nel decorso della malattia e anche in fase di stabilità clinica, grande attenzione e interesse di ricerca sono stati rivolti all’impiego di trattamenti antiinfiammatori volti a limitare la progressione del danno polmonare.
- L’impiego cronico degli steroidi per via sistemica, pur dimostrandosi efficace in termini di ridotto declino funzionale respiratorio e di riduzione della risposta infiammatoria, è stato gravato da effetti collaterali dose-dipendenti inaccettabili che ne hanno precluso l’impiego. D’altra parte non vi sono ancora prove di efficacia a lungo termine dell’impiego di tali farmaci per via inalatoria.
- Esperienze significative sono state riportate con l’ibuprofene che, impiegato cronicamente, si è dimostrato in grado di indurre un rallentamento nel declino funzionale respiratorio, in assenza di effetti collaterali significativi, soprattutto in pazienti di età compresa tra 6 e 17 anni con FEV1 >60%, mentre più scarsi sono i dati in pazienti adulti. Tuttavia, la necessità di monitorare le concentrazioni sieriche del farmaco, che devono rimanere comprese in un range tra 50 e 100 mg/ml, così come il rischio di potenziali effetti collaterali ne hanno limitato l’impiego.
- Esistono dati non conclusivi riguardo al potenziale utilizzo di α1-antitripsina ricombinante e altre molecole ad attività antiproteasica per via aerosolica; attualmente sono tuttavia in fase di sviluppo nuovi inibitori delle proteasi con migliorate caratteristiche farmacologiche che potrebbero rivelarsi di potenziale interesse per il trattamento della fibrosi cistica.
- Sulla scorta dell’esperienza giapponese in merito all’impiego dei macrolidi per il trattamento della panbronchiolite diffusa, sono stati condotti diversi studi per valutarne l’efficacia nei pazienti con FC. Il più ampio di tali studi, che ha valutato l’efficacia dell’azitromicina (somministrata per tre volte/settimana), ha dimostrato un miglioramento del FEV1 e una riduzione delle esacerbazioni respiratorie significativamente maggiore rispetto al placebo nei pazienti con infezione cronica da P. aeruginosa. L’efficacia del farmaco è stata documentata anche nei pazienti senza infezione cronica da P. aeruginosa. Il preciso meccanismo di azione dell’azitromicina non è ancora completamente chiarito; sembrano concomitare nell’effetto sia l’attività antibatterica che, soprattutto, una attività antiinfiammatoria e immunomodulatrice.
- Quando la progressione del danno broncopolmonare determina un quadro di insufficienza respiratoria, l’approccio terapeutico si basa sull’intensificazione della terapia convenzionale, sull’impiego dell’ossigenoterapia ed eventualmente di supporto ventilatorio non invasivo. I progressi nel campo del trapianto polmonare hanno fornito una importante opzione terapeutica per i pazienti con malattia polmonare severa e irreversibile, in cui il trattamento medico convenzionale non è più efficace, che consente di estendere e migliorare la qualità di vita dei pazienti in cui vi sia l’indicazione.
TRATTAMENTO DELLE MANIFESTA ZIONI GASTROENTEROLOGICHE E NUTRIZIONALI
- È particolarmente importante cercare di ottimizzare lo stato nutrizionale nei pazienti con FC; è stato infatti osservato che esso costituisce un importante indice prognostico e che il deficit nutrizionale influenza il declino della funzione respiratoria. Il primo obiettivo è quindi il trattamento dell’insufficienza pancreatica esocrina, che prevede la somministrazione per via orale di enzimi pancreatici a ogni pasto. In presenza di insufficienza pancreatica esocrina, anche se adeguatamente corretta con la terapia sostitutiva, è di comune riscontro il deficit di vitamine liposolubili, per cui è raccomandata la loro supplementazione dopo dimostrazione dell’effettiva carenza. In corso di riacutizzazioni infettive e con il progredire della patologia respiratoria il fabbisogno calorico aumenta mentre diminuisce l’apporto dietetico per l’anoressia; il deficit nutrizionale che ne consegue provoca a sua volta un peggioramento della funzione respiratoria. In queste situazioni occorre interrompere prontamente il circolo vizioso e procedere a· una supplementazione calorica.
- Un ruolo fondamentale riveste il trattamento di alcune delle complicanze più frequenti quale il diabete. La terapia si avvale in qualche caso selezionato degli ipoglicemizzanti orali, ma nella maggior parte dei casi viene impostato precocemente il trattamento insulinico.
- In presenza di cirrosi epatica il trattamento è volto a prevenire le complicanze dell’ipertensione portale. La progressione dell’ipertensione portale con complicanze non responsive alle usuali terapie, così come l’instaurarsi di un deficit di sintesi, costituiscono l’indicazione principale al trapianto cli fegato. In presenza di severa compromissione respiratoria è necessario prendere in considerazione il trapianto fegato-polmone.
TRATTAMENTO EZIOLOGICO DELLA FIBROSI CISTICA
- L’identificazione del gene CFTR e i progressi nella comprensione dei meccanismi biomolecolari attraverso i quali le varie, classi di mutazioni inducono le alterazioni della proteina CFTR hanno portato a una notevole mole di studi e oi investimenti ci ricerca volti a sviluppare trattamenti in grado di correggere il difetto di base della malattia.
- In questo ambito un campo estremamente promettente è quello della terapia genica, che si propone di correggere il difetto di base della malattia trasferendo e facendo esprimere una copia normale del gene CFTR nelle cellule epiteliali delle vie aeree. Sebbene concettualmente semplice, gli studi di terapia genica hanno mostrato numerose difficoltà sotto il profilo pratico e al momento attuale, pur con risultati sperimentali promettenti, la terapia genica, seppur in continua evoluzione, è ancora lontana dall’applicazione clinica. Negli anni più recenti, si sono sviluppate peraltro modalità di terapia genetica molto innovative che mirano a modificare il gene nel suo sito, intervenendo sul punto difettoso, il cosiddetto gene editing. Lo studio in vitro delle possibilità di applicazione di questa tecnica nel campo della FC è tuttavia appena iniziato e non sono ancora noti i problemi e gli ostacoli che incontrerà la sua trasposizione in vivo nell’uomo.
- In questo ambito, tuttavia, l’area di ricerca complessivamente più avanzata e in parte risultata in applicazione clinica è rappresentata dai modulatori di CFTR. Le approfondite conoscenze in merito alle modalità con cui le diverse classi di mutazioni geniche conducono al difetto maturativo o funzionale della proteina CFTR, infatti, hanno portato a un enorme interesse nell’identificazione di molecole in grado di correggere il difetto biomolecolare di base “classe specifico”. In particolare, in questo contesto, una delle strategie terapeutiche più innovative e sviluppate è quella di aumentare l’espressione o la funzione della proteina CFTR mutata con agenti farmacologici: farmaci “correttori” che sono in grado di indurre la maturazione della proteina CFTR mutata e di consentirne l’espressione sulla membrana apicale della cellula (come nel caso della più comune mutazione, la DF508), o farmaci “potenziatori” che agiscono potenziando la funzionalità della proteina presente sulla superficie della cellula ma scarsamente funzionante. Più recentemente sono oggetto di notevole interesse di ricerca i cosiddetti “amplificatori” cioè composti in grado di amplificare la produzione di proteina CFTR in maniera da offrire a correttori e potenziatori maggiore quantità di substrato da recuperare/attivare e gli “stabilizzatori” che contribuiscono a prolungare l’espressione della proteina sulla membrana cellulare e che possono essere utilmente associati ad altri modulatori. Infine sono anche allo studio composti in grado di impedire il blocco prematuro della sintesi di CFTR.
- Il primo tra i modulatori di CFTR entrato nell’uso clinico per un sottogruppo di pazienti, quelli con mutazione G551D, una mutazione di classe III rarissima in Italia, è stato l’Ivacaftor (VX-770), un farmaco potenziatore sviluppato dalla ditta Vertex Pharmaceutics. Nei pazienti con mutazione G551D, il trattamento con questo farmaco per 24 settimane si è dimostrato in grado di indurre un miglioramento della funzionalità respiratoria, di ridurre le esacerbazioni e di indurre aumento di peso, oltre che di normalizzare in media i valori del test del sudore. Successivamente sono state studiate diverse associazioni di correttori e potenziatori, con l’obiettivo in particolare di sviluppare un trattamento che fosse efficace sulla mutazione DF508, la più comune nella razza caucasica. In questo ambito, recentemente è stato sviluppata ed è entrata nell’uso clinico in diversi paesi una triplice associazione di modulatori (elexacaftor/tezacaftor/ivacaftor) che si è dimostrata molto efficace in pazienti di età > 12 anni sia eterozigoti che omozigoti per la mutazione DF508. Notevole interesse di ricerca è anche rivolto all’identificazione e allo studio di molecole attive su canali diversi dal CFTR quali attivatori di TMEM16A (canale del cloro attivato dal calcio) o inibitori del canale del sodio ENaC.
- In conclusione, sebbene attualmente siano stati fatti enormi progressi nel trattamento della fibrosi cistica, che hanno contribuito in maniera determinante al miglioramento della sopravvivenza e della qualità di vita di tali pazienti, è verosimile che in un futuro non lontano gli sforzi della ricerca condurranno alla disponibilità di trattamenti ancora più efficaci, in grado di correggere completamente e in tutti i pazienti il difetto di base della malattia, e pertanto a una reale possibilità di cura della stessa.