Dettagli
- Definizione
- Epidemiologia
- Eziologia e patogenesi
- Anamnesi ed esame obiettivo
- Diagnosi
- Terapia
- Immagine 01
- Immagine 02
- Immagine 03
- Immagine 04
Iperaldosteronismo primario
Definizione
- L’aldosteronismo primario (anche detto Sindrome di Conn) è un disordine delle ghiandole surrenali che provoca un’eccessiva produzione di aldosterone.
- Le cause più comuni sono l’adenoma unilaterale che produce aldosterone e l’iperplasia surrenale bilaterale. Rari casi derivano dal carcinoma surrenale o da condizioni ereditarie di iperaldosteronismo familiare. La produzione inappropriata di aldosterone porta a ritenzione di sodio, ipertensione, complicazioni cardiovascolari, soppressione della renina plasmatica e aumento dell’escrezione di potassio, che a sua volta può portare a ipocaliemia.
Epidemiologia
- Il picco di insorgenza è nei pazienti tra i 40 e i 50 anni di età, senza differenze sostanziali di genere o razza.
Eziologia e patogenesi
- Le 2 cause più comuni includono l’adenoma unilaterale che produce aldosterone (riportato in circa il 50% dei pazienti) e l’iperaldosteronismo idiopatico (iperplasia surrenale bilaterale, in circa il 50% dei pazienti).
- Cause più rare includono l’iperaldosteronismo familiare (FH) che comprende 5 sottotipi che sembrano rappresentare l’1%-5% dei casi di aldosteronismo primario.
- L’FH-1 (chiamato anche aldosteronismo sensibile ai glucocorticoidi) ha un’incidenza di meno dell’1% dei pazienti con aldosteronismo primario. E’ associato ad una fusione della sequenza del promotore del gene CYP11B1 con la regione codificante del gene CYP11B2. Questa variante risponde al trattamento con glucocorticoidi (desametasone a basso dosaggio).
- L’FH-2 è riportato in circa il 5% dei pazienti con aldosteronismo primario ed è associato a mutazioni nel gene CLCN2. Può includere sia l’iperplasia adrenocorticale che l’adenoma produttore di aldosterone (APA) e non risponde al trattamento con glucocorticoidi.
- L’FH-3 è riportato in circa lo 0,3% dei pazienti con aldosteronismo primario ed è associato a mutazioni nel gene KCNJ5. Esso è a sua voltacomposto da 2 sottotipi:
- tipo A: grave, con iperplasia surrenalica bilaterale massiccia, che spesso richiede un intervento chirurgico (surrenalectomia bilaterale);
- tipo B: lieve, con nessuna evidenza di iperplasia surrenalica e buona risposta ai farmaci.
- Il sottotipo FH-4 è molto raro ed associato con una mutazione nel gene CACNA1H.
- L’FH-5 (chiamato anche aldosteronismo primario con convulsioni e anomalie neurologiche [PASNA]) è molto raro, associato a mutazioni germinali nel CACNA1D ela presentazione include sintomi extra surrenali.
- Altre cause più rare includono piccole lesioni iperplastiche di una singola ghiandola surrenale (iperplasia surrenale unilaterale), noduli adrenocorticali multipli unilaterali, adenomi bilaterali che producono aldosterone e carcinomi surrenocorticali.
- Per quanto rigurda la patogenesi, l’interruzione del normale sistema RAAS a causa di una produzione di aldosterone inappropriatamente elevata e relativamente autonoma, può provocare:
- ipertensione dovuta all’espansione di volume e all’attivazione del sistema nervoso simpatico a causa della ritenzione di sodio;
- danni d’organo (come emorragia o infarto cerebrale, infarto miocardico, cardiomegalia, aritmia e insufficienza renale) dovuti a livelli di aldosterone inappropriatamente alti (indipendentemente dal suo effetto sulla pressione sanguigna);
- ipopotassiemia dovuta all’aumento dell’escrezione di potassio;
- alcalosi metabolica dovuta all’aumento dell’escrezione di ioni idrogeno;
- soppressione della renina plasmatica.
- Dei vari tipi di iperaldosteronismo familiare (FH) sappiamo che tutti sono autosomici dominanti. Tra essi ricordiamo l’FH-1 (aldosteronismo sensibile ai glucocorticoidi), causato da una mutazione che crea una fusione tra la regione promotrice del CYP11B1 e le sequenze codificanti del CYP11B2, con conseguente gene chimerico CYP11B1/CYP11B2. La mutazione del gene ibrido risulta in un enzima regolato dall’ormone adrenocorticotropo (ACTH) che sintetizza l’aldosterone (invece dell’angiotensina II e regolato dal potassio). L’espressione dell’aldosterone sintasi avviene nella zona fasciculata invece che nella zona glomulerosa.
- L’FH-2 è causato da mutazioni germinali nel gene del canale del cloruro CLCN2 che aumenta l’efflusso di Cioro. Questo porta a: depolarizzazione della membrana plasmatica, apertura del canale Ca2+ legato al voltaggio, accumulo di Ca2+ citostolico ed attivazione della trascrizione del CYP11B2.
- L’FH-3 è causato da mutazioni germinali in KCNJ5 che determinano una ridotta selettività K+ e un aumento dell’afflusso di Na+ nel citoplasma; questo porta alla depolarizzazione della membrana e all’aumento dei livelli di Ca2+ intracellulare. Inoltre, l’attivazione delle vie legate al Ca2+ innesca la produzione di aldosterone.
- L’FH-4 è causato da mutazioni germinali in CACNA1H che aumenta l’ingresso di Ca2 ed attiva l’espressione di CYP11B2.
- L’FH-5 (aldosteronismo primario con convulsioni e anomalie neurologiche [PASNA]) è causato da mutazioni germinali del canale del calcio nel gene CACNA1D. Esso attiva il potenziale di membrana delle cellule della zona glomerulosa, aumenta l’afflusso di Ca2+ e la produzione di aldosterone e, anche se segue un modello autosomico dominante, la trasmissione è poco probabile.
- Infine, alcune mutazioni germinali in ARMC5 sono state descritte in pazienti con aldosteronismo primario e diverse mutazioni somatiche sono state descritte nell’adenoma che produce aldosterone, anche se ciò non influenza il trattamento.
Anamnesi ed esame obiettivo
- La maggior parte dei pazienti con aldosteronismo primario è asintomatico o presenta un’ipertensione normokalemica. Quest’ultima è spesso resistente al trattamento e pochi casi sono associati a ipopotassiemia (9-37% dei casi) e alcalosi. In caso di ipopotassiemia i sintomi possono includere: fatica, debolezza muscolare e crampi, costipazione (ileo paralitico), paralisi ascendente e insufficienza respiratoria se il potassio sierico < 2 mEq/L (2 mmol/L), poliuria e nicturia (da diabete insipido nefrogenico indotto da ipokaliemia).
- All’esame obiettivo si rileva ipertensione ed in caso di ipopotassiemia i segni possono includere: frequenza cardiaca irregolare con aritmie, diminuzione dei suoni peristaltici, diminuzione dei riflessi tendinei profondi e paralisi ascendente con difficoltà respiratorie se il potassio sierico scende sotto i 2 mEq/L (2 mmol/L).
Diagnosi
- Le indagini diagnostiche dell’aldosteronismo primario comprendono: screening, test di conferma e differenziazione dei sottotipi. Tra i primi ricordiamo che vanno consigliati a tutti i soggetti con: pressione sanguigna sostenuta ≥ 150/100 mm Hg su ciascuna delle 3 misurazioni effettuate in giorni diversi, ipertensione e ipopotassiemia (inclusa quella indotta da diuretici), ipertensione resistente ai farmaci (sopra 140/90 mm Hg e resistente a 3 farmaci antipertensivi convenzionali, uno dei quali deve essere un diuretico) o pressione sanguigna controllata (< 140/90 mm Hg) su ≥ 4 farmaci antipertensivi, ipertensione e incidentaloma surrenale, ipertensione e apnea del sonno, ipertensione e storia familiare di ipertensione precoce o incidente cerebrovascolare all’età < 40 anni ed infine ipertensione e parenti di primo grado con aldosteronismo primario. Va valutata anche la concentrazione plasmatica dell’aldosterone ed il rapporto attività reninica plasmatica (ARR) e, se alto, i test di conferma diventano opportuni (i valori cutoff in vanno da > 20 a > 40, con > 30 che è il più comunemente usato).
- La diagnosi viene confermata quando viene dimostrata una produzione non sopprimibile o autonoma di aldosterone. I pazienti con ARR positiva dovrebbero avere almeno un test di conferma per la diagnosi definitiva, mentre non ce n’è bisogno nei pazienti con ipopotassiemia spontanea, renina plasmatica al di sotto del limite di rilevamento e concentrazione di aldosterone plasmatico > 20 ng/dL. Tra le varie opzioni di test per confermare la diagnosi ricordiamo: il test di carico salino orale, di soppressione salina, di soppressione del fludrocortisone ed il captopril challenge test.
- Il campionamento venoso surrenalico (AVS) è usato per differenziare la malattia unilaterale (adenoma o iperplasia surrenale che produce aldosterone) dalla malattia bilaterale (iperaldosteronismo idiopatico) nei pazienti che saranno sottoposti a surrenalectomia. Se l’aldosteronismo primario è confermato, ulteriori test dovrebbero essere utilizzati per identificarne la causa.
- Tra le indagini più consigliate ricordiamo il controllo del potassio sierico e la concentrazione plasmatica di aldosterone e l’attività plasmatica della renina che vengono utilizzate per calcolare il rapporto aldosterone-renina (ARR). All’ottenere un valore finale alto, un test di conferma è di solito il passo successivo (i cutoff nelle diverse pratiche vanno comunemente da > 20 a > 40, con > 30 che è il più comunemente usato); se invece la concentrazione plasmatica di aldosterone è < 15 ng/dL, vanno considerati i fattori che possono abbassare i livelli di renina, poiché questo può anche aumentare l’ARR.
- Per confermare la diagnosi si utilizzano i test sopra elencati e nessuna ulteriore indagine è richiesta nei pazienti con ipopotassiemia spontanea, attività della renina plasmatica al di sotto del livello di rilevamento e concentrazione di aldosterone plasmatico > 20 ng/dL.
- Al fine di determinare la causa per guidare il trattamento si può eseguire una tomografia computerizzata (TAC) surrenalica per escludere grandi masse che possono rappresentare un carcinoma surrenocorticale e per assistere il radiologo interventista e il chirurgo dove anatomicamente appropriato.
- Quando il trattamento chirurgico è possibile e desiderato dal paziente, il campionamento venoso surrenalico (AVS) dovrebbe essere effettuato per differenziare la malattia bilaterale da quella unilaterale. I pazienti sotto i 35 anni che hanno ipocaliemia spontanea, marcato eccesso di aldosterone e lesioni surrenali unilaterali con caratteristiche radiologiche coerenti con l’adenoma corticale, alla TAC possono non aver bisogno di AVS prima di procedere alla surrenalectomia unilaterale. Test genetici per l’iperaldosteronismo familiare (FH) di tipo I vengono effettuati utilizzando il southern blot o la reazione a catena della polimerasi (PCR) se c’è il sospetto clinico di aldosteronismo che risponde ai glucocorticoidi (insorgenza prima dei 20 anni o storia familiare di aldosteronismo primario o di ictus prima dei 40 anni). In pazienti molto giovani con aldosteronismo primario è consigliato eseguire un test genetico per mutazioni germinali in KCNJ5 risultanti in FH tipo III.
Terapia
- Gli obiettivi del trattamento sono la normalizzazione della pressione sanguigna e dei livelli di potassio e la riduzione dei livelli di aldosterone (che può portare a danni d’organo indipendentemente dalla pressione sanguigna). L’ipertensione va trattata con un approccio al cambiamento nello stile di vita prima dei farmaci.
- Per i soggetti con malattia unilaterale (adenoma produttore di aldosterone o iperplasia surrenale) il trattamento raccomandato consiste nella normalizzazione preoperatoria della pressione sanguigna e dell’ipopotassiemia con spironolattone e integratori orali di potassio. A seguire si raccomanda una surrenalectomia laparoscopica unilaterale con discontinuità della supplementazione di potassio il giorno 1 dopo l’intervento, sospensione dello spironolattone e riduzione della terapia antipertensiva ove possibile. In alternativa si può optare per una terapia con antagonisti del recettore dei mineralocorticoidi nei pazienti che non possono o non vogliono sottoporsi all’intervento chirurgico o nei pazienti con rapporto aldosterone-renina positivo che non possono o non vogliono sottoporsi a ulteriori indagini.
- In caso di adenomi o iperplasie surrenali bilaterali (iperaldosteronismo idiopatico) gli antagonisti del recettore dei mineralocorticoidi sono raccomandati. Lo spironolattone è l’agente preferito e l’eplerenone è un’alternativa.
- Per i pazienti affetti da FH-1 (aldosteronismo sensibile ai glucocorticoidi) il trattamento di scelta è costituito da glucocorticoidi a basso dosaggio per sopprimere la secrezione di ormone adrenocorticotropo ipofisario (ACTH). Negli adulti, la dose di desametasone è dello 0,125-0,25 mg/giorno o prednisone 2,5-5 mg/giorno, assunto al momento di coricarsi. Nei bambini, gli obiettivi di pressione sanguigna dovrebbero allinearsi alle medie specifiche per età e sesso e la dose di glucocorticoidi dovrebbe essere regolata in base all’età e al peso corporeo. Gli antagonisti del recettore dei mineralocorticoidi (spironolattone o eplerenone) possono essere aggiunti come agenti di seconda linea se la pressione sanguigna non è normalizzata con il solo glucocorticoide. Nei pazienti i cui livelli di pressione sanguigna target non vengono raggiunti con desametasone a basso dosaggio, possono essere considerati anche i farmaci antipertensivi standard, come i calcio antagonisti o i beta-bloccanti.
- Per l’FH-2, gli antagonisti del recettore dei mineralocorticoidi (spironolattone o eplerenone) sono la prima scelta.
- In soggetti affetti da FH-3 il tipo A (grave) non risponde ai farmaci e può richiedere una surrenalectomia bilaterale, mentre il tipo B (lieve) può essere tipicamente gestito con antagonisti del recettore dei mineralocorticoidi.
- Per l’FH-4, la terapia non è standardizzata poiché la condizione è molto rara e questa risulta spesso infruttuosa sia con gli antagonisti del recettore dei mineralocorticoidi che con la surrenalectomia unilaterale. Anche i bloccanti dei canali del calcio di tipo T possono essere considerati.
- Per i pazienti con FH-5 (aldosteronismo primario con convulsioni e anomalie neurologiche [PASNA]), i trattamenti riportati includono: bloccanti dei canali del calcio, antagonisti del recettore dei mineralocorticoidi e surrenalectomia se la condizione è lateralizzata
Immagine 01
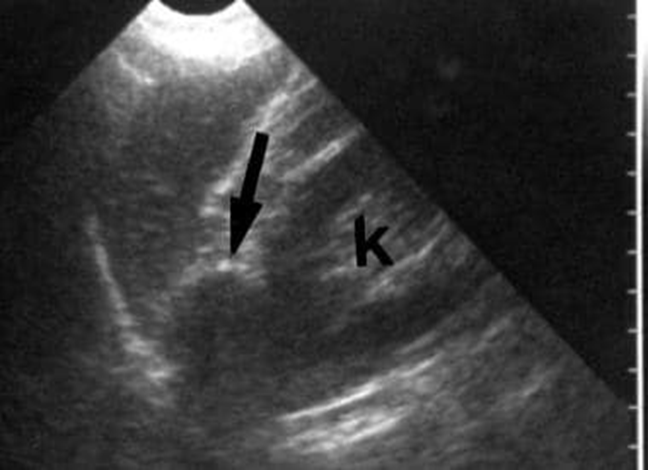
Immagine 01. Ecografia del rene sinistro in paziente donna di 32 anni con ipertensione. La freccia mostra una massa ipoecogena di circa 3 cm superiormente al rene.
Immagine 02
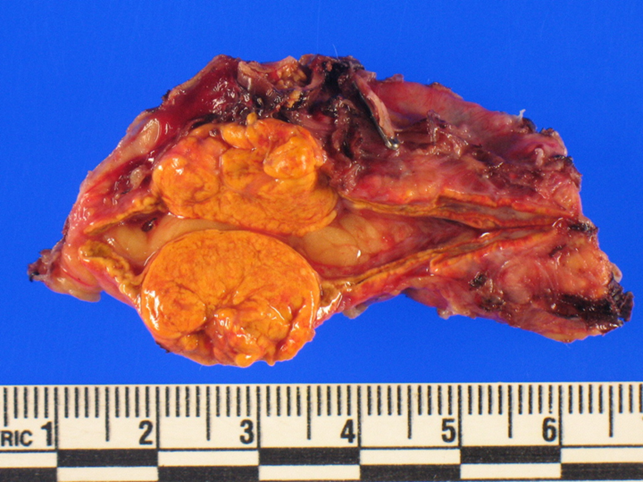
Immagine 02. Tumore soprarenale che produce aldosterone in eccesso come causa della malattia di Conn.
Immagine 03
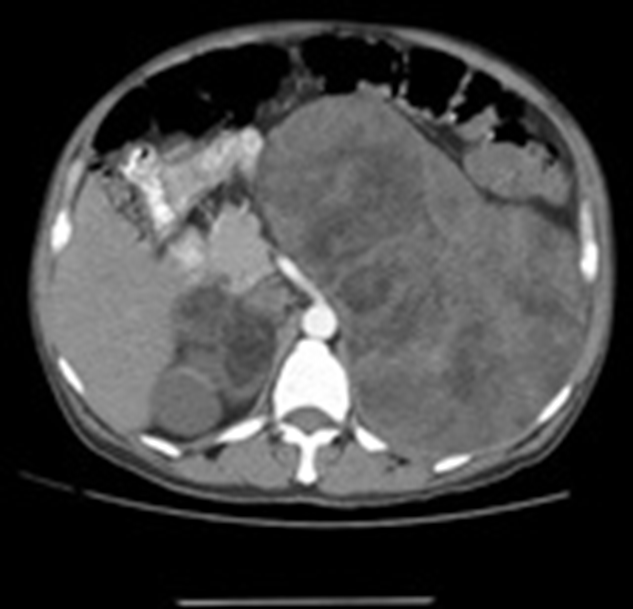
Immagine 03. Mielolipoma surrenalico gigante bilaterale.
Immagine 04
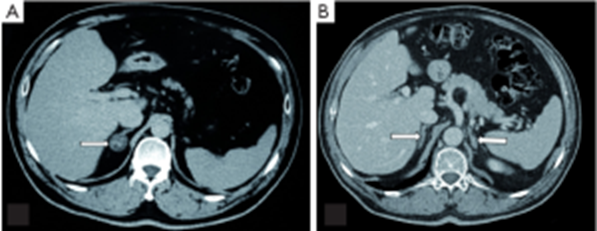
Immagine 04. A: adenoma secernente aldosterone. B: iperplasia surrenalica bilaterale.