Dettagli
- Definizione
- Altrimenti noti come
- Varianti
- Epidemiologia
- Eziologia e patogenesi
- Storia clinica ed esame obiettivo
- Diagnosi
- Gestione
- Immagine 01
- Immagine 02
- Immagine 03
- Immagine 04
Feocromocitoma
Definizione
- I feocromocitomi e i paragangliomi (PPGL) sono tumori neuroendocrini rari, che originano dai gangli simpatici e parasimpatici, a loro volta derivati dalla cresta neurale, e localizzati nella zona midollare del surrene e nei tessuti adiacenti la catena ortosimpatica paravertebrale:
- il feocromocitoma è un tumore localizzato nella midollare del surrene che frequentemente causa secrezione di catecolamine, prodotte dalle cellule cromaffini;
- con il termine paraganglioma si intende un insieme di neoplasie extra-surrenali, che possono o meno secernere catecolamine:
- i paragangliomi ortosimpatici sono tumori che secernono catecolamine e sono solitamente localizzati nel retroperitoneo (meno comunemente nella pelvi o nel torace);
- i paragangliomi parasimpatici sono frequentemente localizzati nella base cranica e nel collo, e solo il 5% è in grado di secernere catecolamine.
- la maggior parte dei feocromocitomi e paragangliomi dimostrano caratteri di benignità:
- le metastasi sono riscontrate nel:
- 5%-20% dei feocromocitomi;
- fino al 35% dei paragangliomi ortosimpatici (il rischio varia a seconda della localizzazione e tende ad aumentare in caso di paragangliomi mediastinici e addominali)
- i siti delle ripetizioni tumorali più comuni includono ossa, polmoni, fegato e linfonodi.
- le metastasi sono riscontrate nel:
Altrimenti noti come
- Tumori cromaffini – le neoplasie che secernono catecolamine (feocromocitoma e paraganglioma ortosimpatico) presentano un caratteristico colore marrone scuro quando vengono trattate con sali di cromo.
- “Tumore cromaffine adrenergico” con cui ci si riferisce al feocromocitoma.
Varianti
- I feocromocitomi e i paragangliomi possono essere classificati in forme familiari (ereditarie) o sporadiche:
- circa il 35%-40% di tutti i feocromocitomi e paragangliomi negli adulti sono associati a condizioni di predisposizione ereditaria;
- questa predisposizione si verifica in circa l’80% dei casi pediatrici;
- i feocromocitomi familiari sono associati a mutazioni della linea germinale (mutazioni riscontrabili in tutte le cellule dell’organismo) e che possono essere condivise con sindromi tumorali ereditarie, quali le neoplasie endocrine multiple di tipo 2 (MEN2), la malattia di von Hippel-Lindau o la neurofibromatosi di tipo 1;
- la predisposizione familiare per il paraganglioma si associa invece a mutazioni autosomiche dominanti, localizzate nel complesso II della succinato deidrogenasi (SDHD). Le mutazioni possono verificarsi in qualsiasi subunità del gene (SDHA, SDHB, SDHC, SDHD) o dei cofattori, tuttavia, mutazioni loss-of-function nella subunità SDHB sono più comunemente associate ai feocromocitomi;
- i feocromocitomi e i paragangliomi sporadici non sembrano associati ad alcuna mutazione genetica nota; tuttavia, una percentuale sempre crescente di pazienti (fino al 40% nel 2019) manifesta mutazioni germinali, in assenza di anamnesi familiare rilevante.
- I tumori funzionanti (feocromocitoma e paraganglioma ortosimpatico) sintetizzano e secernono catecolamine e loro metaboliti:
- i sottotipi includono:
- tumori noradrenergici, che secernono norepinefrina e normetanefrina;
- tumori adrenergici, che secernono epinefrina, norepinefrina, metanefrina e normetanefrina:
- la norepinefrina è la catecolamina predominante sintetizzata e secreta dalle cellule gangliari e dai neuroni adrenergici;
- la secrezione di epinefrina è in gran parte esclusiva dei feocromocitomi intra-surrenali.
- tumori dopaminergici (rari), che secernono dopamina, acido omovanillico e metossi-tiramina.
- l’80%-85% di queste lesioni è rappresentato da feocromocitomi;
- il 15%-20% da paragangliomi ortosimpatici.
- i sottotipi includono:
- Le neoplasie non funzionanti (paraganglioma parasimpatico) non sintetizzano o secernono catecolamine, quindi sono comunemente individuate incidentalmente durante indagini di imaging o poiché causano effetto massa sulle strutture adiacenti.
Epidemiologia
- La malattia sporadica si presenta comunemente in soggetti di 40-50 anni, ma può verificarsi in tutte le età.
- La malattia familiare è solitamente riconosciuta in pazienti di età inferiore ai 40 anni, con una sempre più precoce diagnosi, probabilmente grazie allo screening genetico delle condizioni associate.
- Il 10%-20% dei feocromocitomi e dei paragangliomi sono diagnosticati nei bambini:
- più comune negli adolescenti;
- età media alla diagnosi 11-13 anni.
Eziologia e patogenesi
Cause
- Entrambe le neoplasie possono avere presentazione sporadica o familiare.
- Sono associate a mutazioni identificate nei pazienti con sindromi familiari di paraganglioma-fecromocitoma (tutte ereditate in modo autosomico dominante con il caratteristico imprinting materno che interessa il gene SDHAF2).
- Esempi di ulteriori geni associati all’insorgenza del feocromocitoma includono:
- KIF1B (membro della famiglia delle chinesine 1B);
- EGLN1 (fattore ipossia-inducibile 1 della famiglia egl-9);
- FH (fumarato idratasi);
- Sindromi associate al paraganglioma senza una chiara causa genetica:
- triade di Carney – paraganglioma ortosimpatico extra-surrenale, sarcoma stomale gastrico e condroma polmonare (adenoma corticale surrenale e leiomioma esofageo possono anche associarsi)
- diade Carney-Stratakis – paragangliomi e tumori stromali gastrointestinali:
- sembra avere una trasmissione autosomica dominante con penetranza incompleta;
- mutazioni dei geni SDHB, SDHD e SDHC sono state identificate in alcuni pazienti.
- paragangliomi associati a policitemia e angiomi retinici con mutazione somatica di HIF-2alpha.
Patogenesi
- I tumori che secernono catecolamine (feocromocitomi e paragangliomi ortosimpatici) derivano dal tessuto cromaffine presente nella midollare del surrene o dai gangli simpatici extra-surrenali:
- localizzazione dei tumori:
- 80%-85% origina dalla midollare del surrene;
- il 10%-20% deriva dal tessuto cromaffine extra-surrene, localizzato nell’addome, nel torace o nella pelvi.
- l’eccesso, spesso episodico, di produzione e/o secrezione di catecolamine (incluse epinefrina, norepinefrina e dopamina) da parte del tumore, è causa della maggior parte dei segni e sintomi (tra cui ipertensione, cefalea, sudorazione e tachicardia).
- localizzazione dei tumori:
- I paragangliomi parasimpatici sono di solito non secernenti e derivano da cellule neuroendocrine dei gangli extra-surrenali:
- comunemente localizzati alla base del cranio o nel collo;
- la sintomatologia tipica è secondaria all’effetto massa, dal momento che il 95% non secerne ormoni, che dipende principalmente dalla sede del tumore.
Storia clinica ed esame obiettivo
Presentazione clinica
- Il feocromocitoma e il paraganglioma sono spesso asintomatici e si presentano come:
- massa surrenale o retroperitoneale identificata incidentalmente durante indagini radiologiche eseguite per ragioni non correlate;
- anomalie biochimiche o radiologiche riscontrate durante lo screening di soggetti a rischio per patologie genetiche.
- Possono essere unilaterali o bilaterali.
- I tumori bilaterali possono svilupparsi in modo sincrono o anche a distanza di oltre 10 anni, specialmente nei pazienti con mutazioni germinali RET1.
- La presentazione clinica tipica negli adulti è dovuta all’eccesso sintomatico di catecolamine:
- la triade classica comprende
- cefalea parossistica (riportata nel 60%-90% dei casi);
- sudorazione profusa (riferita dal 55%-75% dei pazienti);
- palpitazioni (riportate nel 50%-70% dei casi).
- altri segni frequenti possono essere:
- ipertensione, a carattere:
- sostenuto (riportato nel 50% dei casi);
- parossistico (nel 30%);
- ortostatico (nel 12%).
- panico o ansia (riferita dal 20%-40% dei soggetti);
- aritmie.
- ipertensione, a carattere:
- segni e i sintomi meno comuni possono includere:
- pallore (40% dei casi);
- calo ponderale (nel 20%-40% dei casi);
- febbre (nel 60%);
- astenia (nel 25%-40%);
- iperglicemia (nel 40%);
- nausea o vomito.
- la triade classica comprende
- La presentazione clinica può riflettere la marcata secrezione norepinefrina rispetto a quella di epinefrina da parte della neoplasia:
- i tumori che secernono prevalentemente norepinefrina sono spesso associati a un’ipertensione sostenuta;
- i tumori che secernono prevalentemente epinefrina spesso causano ipertensione parossistica ed episodi di ipotensione ortostatica.
- I feocromocitomi possono anche causare esiti cardiaci di lunga durata, come:
- cardiomiopatia dilatativa o ipertrofica;
- insufficienza cardiaca.
- Diversa invece appare la presentazione clinica negli adulti con paraganglioma parasimpatico (per lo più non funzionante), che risulta essere associata all’effetto massa:
- tra i segni più comuni, la presenza una massa cervicale indolore a lento accrescimento, anomalie dei nervi cranici, cambiamenti di voce e deficit dell’udito;
- i casi severi presentano comunemente deficit dei nervi cranici inferiori;
- i sintomi possono suggerire la localizzazione della massa tumorale:
- tinnito, ipoacusia e altri deficit dei nervi cranici possono suggerire un paraganglioma giugulo-timpanico;
- massa nella regione del collo, raucedine, disfagia, disfonia, dolore, tosse e ab ingestis possono suggerire un paraganglioma vagale;
- la massa laterale del collo e i deficit dei nervi cranici possono suggerire un paraganglioma del corpo carotideo.
- Presentazione clinica nei bambini:
- i bambini con tumori secernenti hanno più probabilità di manifestare un’ipertensione sostenuta rispetto agli adulti (riportata nel 60%-90% dei casi);
- segni tipici possono includere:
- episodi parossistici della classica triade (cefalea, palpitazioni e sudorazione profusa);
- pallore;
- ipotensione e sincope;
- tremore;
- ansia;
- sintomi non specifici, come:
- visione offuscata;
- dolore addominale;
- diarrea e altri sintomi gastrointestinali;
- perdita di peso;
- iperglicemia;
- poliuria e polidipsia;
- febbre non superiore ai 38°C;
- deficit neurocomportamentali o declino del rendimento scolastico.
- la presentazione clinica dei tumori parasimpatici non secretori può includere:
- perdita dell’udito;
- tinnito;
- altri sintomi causati dalla compressione estrinseca, come
- raucedine;
- pienezza faringea;
- disfagia;
- tosse;
- dolore.
Storia clinica
- I sintomi parossistici possono persistere per alcuni minuti fino a 1 ora e sono spesso stimolati da:
- anestesia;
- cibo;
- minzione (in caso di paraganglioma della vescica);
- cambiamento posizionale;
- esercizio fisico;
- composti chimici o droghe.
- Le donne incinte possono presentare sintomi parossistici, scatenati da:
- contrazioni uterine;
- utero in crescita;
- movimento fetale;
- parto;
- fattori farmacologici come l’anestesia;
- compressione del tumore da parte dell’utero gravido in posizione supina.
- Indagare su eventuali episodi di ansia o di attacchi di panico, specialmente se episodici:
Anamnesi farmacologica
- Ricercare possibili correlazioni temporali tra l’insorgenza dei sintomi e l’assunzione di farmaci come:
- agonisti dei recettori della dopamina (compresi alcuni agenti antiemetici e antipsicotici);
- agonisti dei recettori beta-adrenergici;
- simpaticomimetici;
- analgesici oppioidi;
- inibitori della ricaptazione della noradrenalina o antidepressivi triciclici;
- inibitori della monoammina ossidasi;
- corticosteroidi;
- agenti bloccanti neuromuscolari;
- peptidi;
- inibitori della ricaptazione della serotonina (riportati raramente).
- Indagare sull’uso di farmaci che potrebbero interferire con l’analisi diagnostica e falsare i risultati di test biochimici sul plasma o sulle urine, tra cui:
- acetaminofene;
- alfa-metildopa;
- buspirone;
- fenossibenzamina;
- antidepressivi triciclici;
- inibitori della monoammino-ossidasi (MAO);
- levodopa;
- simpaticomimetici (incluse anfetamine, efedrina);
- sulfasalazine;
- decongestionanti o altri farmaci contenenti agonisti dei recettori adrenergici;
- agenti antipsicotici;
- droghe illecite, come la cocaina o l’eroina, o l’astinenza da queste.
Anamnesi patologica remota
- Chiedere di condizioni che potrebbero suggerire sindromi genetiche associate, tra cui:
- malattia di Von Hippel-Lindau (VHL):
- carcinoma renale a cellule chiare o cisti;
- emangioblastomi del sistema nervoso centrale (SNC) e della retina;
- tumori o cisti pancreatiche;
- tumori endolinfatici;
- cisti epididimali.
- sindrome da neoplasia endocrina multipla 2A o 2B:
- carcinoma midollare della tiroide;
- iperparatiroidismo;
- ganglioneuromi;
- habitus marfanoide.
- neurofibromatosi di tipo 1:
- neurofibromi;
- tumori del sistema nervoso centrale e periferico.
- sindromi ereditarie di paraganglioma-fecromocitoma dovute a mutazioni del gene SDH, che raramente possono includere:
- tumori stromali gastrointestinali;
- carcinoma renale a cellule chiare.
- malattia di Von Hippel-Lindau (VHL):
Esame obiettivo
Generale
- Ricercare la presenza di ipertensione arteriosa (cronica o episodica):
- circa il 50% degli adulti con feocromocitoma presenta un’ipertensione spesso farmaco-resistente;
- circa il 30% degli adulti con feocromocitoma presenta un’ipertensione parossistica/episodica;
- circa il 60%-90% dei bambini manifesta ipertensione sostenuta;
- Anche l’ipotensione ortostatica/posizionale potrebbe essere presente:
- può verificarsi in pazienti con ipertensione sostenuta e meno comunemente in soggetti che presentano ipertensione parossistica;
- attribuita alla down-regolazione dei recettori alfa adrenergici a causa degli elevati valori di catecolamine e della diminuzione del volume ematico.
Cutaneo
- Indagare l’eventuale pallore cutaneo (riportato da circa il 40% dei pazienti con feocromocitoma).
- L’iperpigmentazione della cute (macchie cafe-au-lait, lentiggini ascellari e inguinali) può suggerire la neurofibromatosi di tipo 1.
- Lesioni iperpigmentate possono verificarsi anche in pazienti con neoplasia endocrina multipla di tipo 2A e lichen amiloidosico cutaneo.
- Neuromi della mucosa, che tipicamente coinvolgono le labbra e la lingua, possono suggerire una neoplasia endocrina multipla di tipo 2B.
Regione del collo
- Valutazione per il paraganglioma della testa e del collo:
- ispezionare la base del cranio e il collo;
- la presenza di una massa con diametro maggiore cranio-caudale, associata ad un fremito palpabile con localizzazione in prossimità della biforcazione dell’arteria carotide nella parte supero-laterale del collo, può suggerire la diagnosi di paraganglioma del corpo carotideo;
- una massa di colore blu, pulsante, dietro la membrana timpanica intatta può suggerire un paraganglioma giugulo-timpanico;
- deficit dell’udito;
- deficit dei nervi cranici.
Cardiaco
- Ricercare la presenza di eventuale tachicardia (anche parossistica) o di aritmie.
- Valutare segni di cardiomiopatia ipertrofica.
Respiratorio
- Possono apprezzarsi crepitii/rantoli se presente edema polmonare.
Addominale
- Ispezionare e palpare eventuali masse addominali.
Muscoloscheletrico
- Valutare le anomalie muscolo-scheletriche associate alla neoplasia endocrina multipla di tipo 2B, tra cui
- habitus marfanoide;
- cifoscoliosi;
- lordosi;
- lassità articolare.
Diagnosi
- Sospettare un feocromocitoma o un paraganglioma in pazienti con una presentazione clinica caratterizzata da una delle seguenti:
- ipertensione parossistica o resistente alla farmacoterapia;
- crisi ipertensive durante interventi chirurgici o a seguito di anestesia;
- riscontro di incidentaloma surrenalico con un’attenuazione > 10 unità Hounsfield sulla TAC senza contrasto;
- riferiti attacchi di panico improvvisi;
- storia familiare di sindrome genetica associata a PPGL;
- esacerbazione della sintomatologia a seguito dell’assunzione di farmaci noti come fattori precipitanti la clinica in pazienti affetti da PPGL.
- I PPGL asintomatici sono spesso diagnosticati incidentalmente in pazienti con:
- riscontro incidentale di una massa surrenale o retroperitoneale;
- risultato anomalo di un test biochimico o di imaging durante il monitoraggio di un soggetto che presenta un rischio genetico o durante lo screening di un membro della famiglia a rischio.
- La diagnosi definitiva di feocromocitoma o paraganglioma ortosimpatico si basa sulla positività del test biochimico per la ricerca di un eccessivo rilascio di catecolamine e l’individuazione radiologica del tumore:
- le metanefrine plasmatiche o le metanefrine frazionate urinarie (raccolte nelle urine delle 24 ore) sono considerate il gold standard e dovrebbero essere eseguite come esame di prima linea;
- le metanefrine plasmatiche e le metanefrine frazionate urinarie sembrano più sensibili per la diagnosi di feocromocitoma rispetto alla catecolamina plasmatica o ad altri test urinari;
- la raccolta di campioni di plasma con il paziente in posizione supina può avere una maggiore sensibilità rispetto alla raccolta di campioni di plasma con il paziente in posizione seduta e una maggiore specificità rispetto agli studi sulle urine di 24 ore per la diagnosi di feocromocitoma e paraganglioma;
- il test di soppressione della clonidina può aiutare a discernere i risultati falsi positivi dai falsi negativi ottenuti al test della metanefrina o nella valutazione dei pazienti con livelli borderline di catecolamina e metanefrina;
- eseguire studi di imaging per la localizzazione del tumore solo dopo aver eseguito diagnosi biochimica:
- eseguire una tomografia computerizzata o una risonanza magnetica (MRI) addominale;
- se l’imaging addominale è negativo, considerare la risonanza magnetica della base cranica, del collo, del torace e della pelvi;
- a causa dell’aumento della frequenza delle patologie neoplastiche o di malattie metastatiche, se la massa è grande (> 6 cm di diametro) o extra-surrenale, multifocale (come può comunque avvenite nei paragangliomi della base cranica e del collo), o ricorrente, considerare l’esecuzione di imaging funzionale utilizzando uno dei seguenti:
- F-fluorodeossiglucosio (F-FDG)-PET/CT;
- Scintigrafia con metaiodobenzilguanidina iodo-marcata (I-MIBG);
- Ga-DOTATATE PET/CT;
- La diagnosi di malattia metastatica si basa sull’evidenza di un tumore in siti dove le cellule cromaffini non sono normalmente presenti (nessun test patologico affidabile può differenziare tra tumore primario e uno metastatico).
- Una volta stabilita la diagnosi, eseguire test genetici per rilevare la mutazione specifica e guidare la gestione e il follow-up
Gestione
- Raccomandazioni generali:
- si raccomanda un approccio personalizzato a causa della variabilità delle presentazioni genotipo-fenotipo del feocromocitoma ereditario e del paraganglioma;
- i pazienti dovrebbero essere valutati da team multidisciplinari in centri specializzati (soprattutto donne in gravidanza e pazienti con malattia metastatica).
- Prima dell’intervento, la gestione medica preoperatoria è fondamentale per controllare i sintomi causati dall’increzione di catecolamine, stabilizzare la pressione arteriosa e la frequenza cardiaca nonché per prevenire complicazioni pericolose per la vita (comprese le crisi ipertensive) durante l’intervento:
- iniziare il trattamento medico preoperatorio almeno 7-14 giorni prima dell’intervento, per consentire un’adeguata normalizzazione della pressione arteriosa e della frequenza cardiaca:
- il blocco adrenergico preoperatorio è raccomandato in tutti i pazienti con feocromocitoma e paraganglioma funzionanti poiché il rilascio di elevati livelli di catecolamine durante l’intervento potrebbe causare crisi ipertensive e aritmie;
- usare alfa-bloccanti per la gestione iniziale:
- la fenossibenzamina è un alfa-antagonista non selettivo e irreversibile ed è spesso usato come terapia iniziale per via dell’irreversibilità dell’effetto; tuttavia, il suo utilizzo può essere limitato dalla scarsa biodisponibilità, dagli effetti avversi e dal costo elevato;
- alternative alla fenossibenzamina includono gli antagonisti selettivi alfa-1-adrenergici come la prazosina, doxazosina o terazosina.
- i beta-bloccanti possono essere utilizzati per il controllo della tachicardia solo dopo aver raggiunto un ottimale controllo della pressione arteriosa con gli alfa-bloccanti (i beta-bloccanti non devono essere utilizzati in assenza di alfa-bloccanti a causa della loro scarsa efficacia nel bloccare la stimolazione dei recettori alfa adrenergici che porta alla crisi ipertensiva);
- considerare l’utilizzo di calcio-antagonisti nei pazienti con un controllo inadeguato della pressione sanguigna con i soli alfa-bloccanti, o come alternativa agli alfa-bloccanti nei pazienti con gravi effetti collaterali (la monoterapia con i calcio-antagonisti può anche essere considerata nei pazienti con pressione sanguigna normale o ipertensione preoperatoria molto lieve);
- mentre gli obiettivi cardiovascolari ottimali devono ancora essere definiti, i seguenti valori sono considerati accettabili:
- pressione < 130/80 mmHg in posizione supina e pressione sistolica > 90 mm Hg in posizione eretta;
- frequenza cardiaca tra i 60-70 battiti al minuto da seduti e 70-80 battiti al minuto in piedi.
- l’inizio di una dieta ad alto contenuto di sodio (per esempio 5.000 mg/giorno) con ampio apporto di liquidi (per esempio 2,5 L/giorno) dovrebbe essere usato in combinazione con il blocco adrenergico prima dell’intervento.
- iniziare il trattamento medico preoperatorio almeno 7-14 giorni prima dell’intervento, per consentire un’adeguata normalizzazione della pressione arteriosa e della frequenza cardiaca:
- La chirurgia è il trattamento di scelta nei pazienti con malattia non metastatica o metastasi resecabili:
- le opzioni chirurgiche includono la surrenalectomia totale o la surrenalectomia con risparmio della corticale (surrenalectomia parziale), entrambe le quali possono essere eseguite tramite approcci laparoscopici o aperti;
- la surrenalectomia minimamente invasiva (come la surrenalectomia laparoscopica) è raccomandata per la maggior parte dei feocromocitomi;
- la chirurgia aperta può essere necessaria nei pazienti con feocromocitomi grandi (per esempio, > 6 cm) o invasivi;
- la chirurgia aperta è tipicamente eseguita per i paragangliomi a causa della loro maggiore probabilità di metastatizzare e per la peculiare localizzazione in aree difficilmente accessibili laparoscopicamente;
- la chirurgia laparoscopica è comunque una valida opzione per piccoli paragangliomi non invasivi localizzati in sedi facilmente aggredibili.
- Gestione nei bambini:
- come negli adulti, la gestione nei bambini comporta una preparazione preoperatoria con alfa-bloccanti, beta-bloccanti e altri farmaci, così come l’espansione del volume per controllare l’ipertensione, l’aritmia e prevenire le complicazioni di una crisi ipertensiva prima della gestione definitiva con la chirurgia;
- l’obiettivo da raggiungere per la pressione arteriosa è < 50° percentile per età e altezza;
- la resezione laparoscopica e le procedure che risparmiano la corticale surrenale (surrenalectomia parziale) sono tipicamente preferite nei bambini, specialmente nei bambini con malattia surrenale bilaterale.
- Gestione nelle donne incinte:
- la gestione medica preoperatoria è necessaria per ridurre al minimo il rischio di gravi complicazioni dovute al rilascio massiccio di catecolamine:
- si raccomanda una dieta ad alto contenuto di sodio e una sufficiente assunzione di liquidi per invertire la contrazione del volume sanguigno indotta dalla catecolamina prima dell’intervento e prevenire una grave ipotensione dopo l’asportazione del tumore;
- la gestione medica include alfa-bloccanti, beta-bloccanti, calcio-antagonisti o labetalolo;
- gli alfa-bloccanti come la fenossibenzamina e la doxazosina possono attraversare la placenta ed essere trasferiti nel latte materno, quindi si raccomanda il monitoraggio del neonato per i primi 3 giorni postnatali;
- considerare come target di pressione sanguigna valori di 140/90 mmHg.
- la gestione chirurgica varia in base al grado di avanzamento della gravidanza al momento dell’individuazione del tumore:
- considerare la rimozione endoscopica del tumore prima delle 24 settimane di gestazione, in quanto il rischio di aborto spontaneo è più basso (l’approccio transperitoneale con paziente in posizione di decubito laterale è preferito per la rimozione endoscopica del tumore);
- rinviare la rimozione del tumore fino a o dopo il parto se diagnosticato e rilevato nel terzo trimestre, e considerare il trattamento medico per gestire l’eccesso di catecolamina fino ad allora:
- il parto cesareo è da preferire in quanto associato a meno complicanze in queste situazioni;
- tra le strategie è possibile includere la rimozione della massa neoplastica in concomitanza al parto cesareo o il rinvio dell’intervento per diverse settimane dopo il parto.
- la gestione medica preoperatoria è necessaria per ridurre al minimo il rischio di gravi complicazioni dovute al rilascio massiccio di catecolamine:
- Gestione del feocromocitoma e del paraganglioma metastatici:
- fornire cure di supporto preoperatorie per alleviare i sintomi legati all’eccesso di catecolamine e ridurre il rischio di crisi ipertensive perioperatorie;
- considerare la resezione citoriduttiva per ridurre il carico tumorale e controllare l’ipersecrezione ormonale;
- la radioterapia con vettori radiomarcati mirati, come lo iodio 131-metaiodobenzilguanidina (131I-MIBG) è un’opzione per i pazienti con un carico tumorale elevato o una malattia non resecabile con un buon assorbimento del 123I-MIBG.
- Garantire un follow-up a lungo termine con monitoraggio dei livelli di catecolamina per più di 10 anni dopo un intervento chirurgico eseguito per un feocromocitoma/paraganglioma non metastatico:
- considerare la misurazione dei livelli di metanefrina nel plasma o nelle urine durante il follow-up per identificare una malattia persistente;
- considerare test biochimici annuali per tutta la vita volti ad identificare una malattia metastatica o ricorrente.
Immagine 01
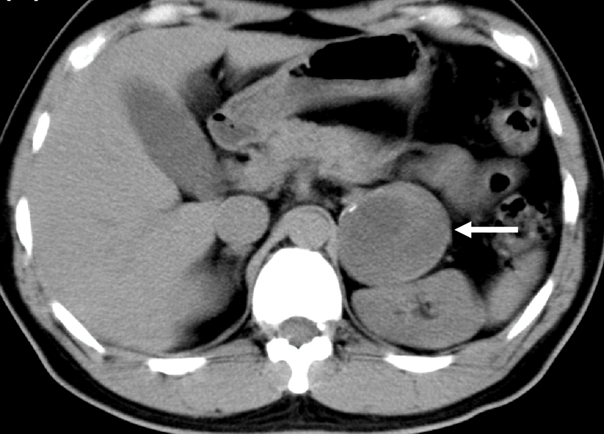
Immagine 01. Scansione assiale di tomografia computerizzata in un uomo di 32 anni, che dimostra la presenza di un incidentaloma surrenalico di 6 cm con calcificazioni periferiche.
Immagine 02
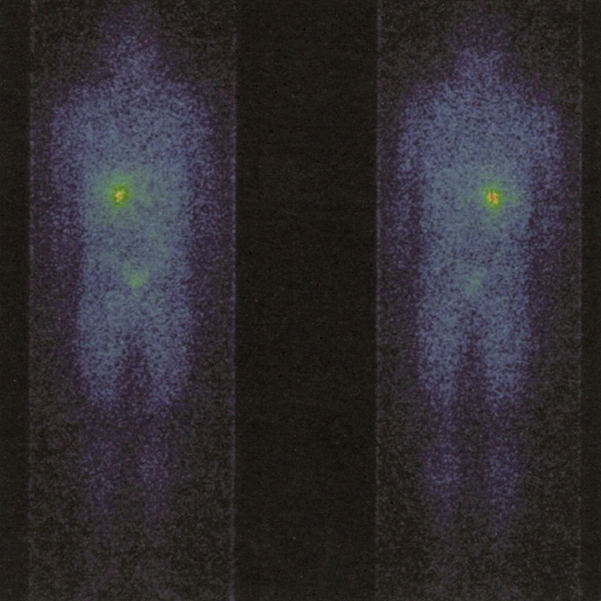
Immagine 02. Scintigrafia con metaiodobenzilguanidina marcata con iodio che dimostra la presenza di un accumulo focale di contrasto nella ghiandola surrenale di destra. Assenti evidenze di metastasi o di feocromocitoma extra-surrenalico.
Immagine 03
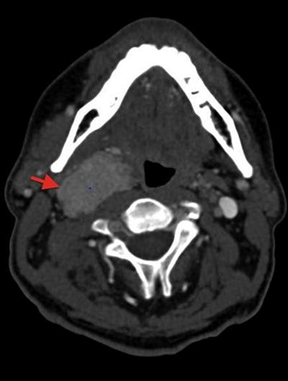
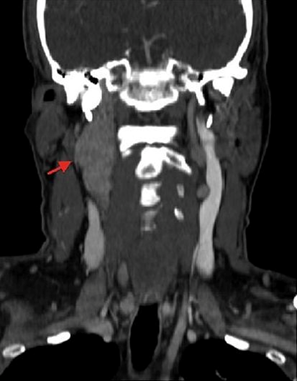
Immagine 03. Immagini TAC in un paziente maschio in piena salute di 39 anni. A) Scansione assiale a livello del corpo vertebrale di C2, che dimostra la presenza di un paraganglioma della carotide di destra. B) Sezione coronale in cui si apprezza l’estensione cranio-caudale della neoplasia che decorre lungo il segmento superiore della carotide comune.
Immagine 04
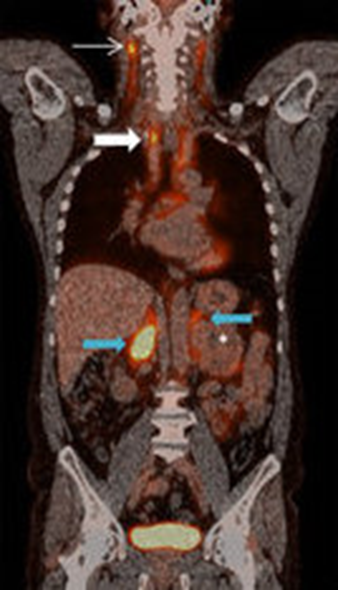
Immagine 04. Sezione coronale di PET/TC in un paziente maschio di 55 anni, che dimostra la presenza di un feocromocitoma sporadico del surrene sinistro. Il tumore esibisce un avido uptake del mezzo di contrasto (18 F-FDG), normalmente anche captato dalle urine e dal grasso localizzato nel mediastino e nel collo.