Dettagli
- Sintesi
- Descrizione
- Chiamato anche
- Definizioni
- Tipi
- Epidemiologia
- Eziologia e patogenesi
- Storia e fisica
- Diagnosi
- Gestione
- Complicazioni e prognosi
- Prevenzione e screening
- Definizione
- Informazioni generali
- Epidemiologia
- Eziologia e patogenesi
- Storia e fisica
- Diagnosi
- Trattamento
- Complicazioni e prognosi
- Panoramica e raccomandazioni
- Epidemiologia
- Eziologia e patogenesi
- Storia e fisica
- Esame obiettivo
- Diagnosi
- Trattamento
- Complicazioni e prognosi
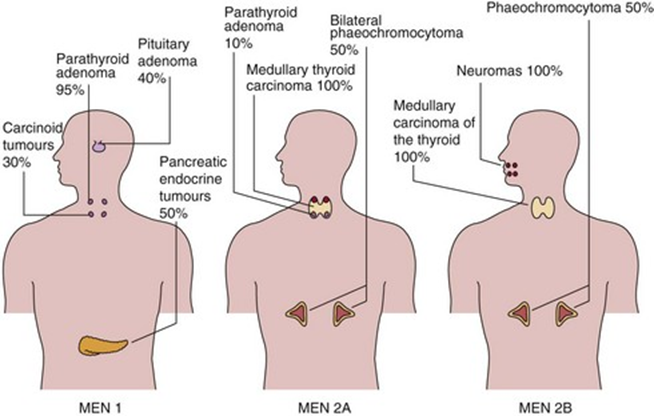
Neoplasia-endocrina
MEN1
Sintesi
- La neoplasia endocrina multipla di tipo 1 (MEN1) è una patologia a trasmissione autosomico dominante e caratterizzata dallo sviluppo di specifici tumori endocrini, principalmente delle paratiroidi, dell’ipofisi anteriore e tumori delle isole pancreatiche, anche se possono insorgere altri tipi di tumori neuroendocrini o non endocrini.
- La MEN 1 è causata da mutazioni germinali del gene oncosoppressore MEN1 situato sul cromosoma 11q13.1.
- Le caratteristiche classiche della MEN 1 e le manifestazioni tumorali più comuni includono:
- adenoma paratiroideo che si manifesta generalmente con un quadro di iperparatiroidismo primario (quindi nefrolitiasi, nefrocalcinosi, disfunzione renale, demineralizzazione ossea);
- adenoma ipofisario che si manifesta con iperprolattinemia (galattorrea, infertilità), aumentata secrezione degli ormoni ipofisari (acromegalia, gigantismo, malattia di Cushing, ipertiroidismo centrale, tumori gonadotrofi secernenti), ma anche possibile insufficienza ipofisaria ed effetto massa;
- tumori neuroendocrini pancreatici (NETs) che si manifestano con ulcere peptiche (nel caso del gastrinoma), ipoglicemia (in caso di insulinoma), perdita di peso, eruzione cutanea (in caso di glucagonoma), grave diarrea acquosa (in caso di VIPoma, gastrinoma);
- manifestazioni cutanee come lipomi, angiofibromi, collagenomi.
- I tumori legati alla MEN1 hanno un potenziale di malignità e i pazienti con MEN 1 sono a rischio di morte prematura.
Descrizione
- La neoplasia endocrina multipla di tipo 1 (MEN1) è una malattia a trasmissione autosomica dominante caratterizzata dallo sviluppo di tumori endocrini, tra cui tumori delle paratiroidi, dell’ipofisi anteriore, tumori delle isole pancreatiche (più comune), tumori delle ghiandole surrenali e tumori del sistema neuroendocrino dello stomaco, duodeno, bronchi e timo (comune).
- La MEN1 può anche essere associata a tumori non endocrini, compresi collagenomi, lipomi, meningiomi, ependimomi, leiomiomi e angiofibromi facciali.
Chiamato anche
- MEN1
- MEN I
- MEN tipo 1
- MEN tipo I
- Sindrome MEN-1
- Adenomatosi endocrina multipla
- Sindrome di Wermer
Definizioni
- La MEN 1 differisce da:
- Neoplasia endocrina multipla di tipo 2 (MEN 2)
- MEN tipo 2A (anche detta sindrome di Sipple), rappresenta il 70%-95% dei pazienti con MEN 2. Si manifesta con:
- carcinoma midollare della tiroide (carcinoma midollare familiare riconosciuto come variante di MEN2A);
- feocromocitoma;
- iperparatiroidismo primario;
- e può associarsi a:
- lichen amiloidosico cutaneo (chiamato anche lichen planus amiloidosico);
- Malattia di Hirschsprung.
- MEN tipo 2B, costituisce circa il 5%-10% dei pazienti con MEN 2, occasionalmente è anche detto MEN3. Si manifesta con:
- carcinoma midollare della tiroide;
- feocromocitoma;
- e può associarsi a
- ganglioneuromatosi;
- habitus dei marfanoidi.
- MEN tipo 2A (anche detta sindrome di Sipple), rappresenta il 70%-95% dei pazienti con MEN 2. Si manifesta con:
- La neoplasia endocrina multipla di tipo 4 caratterizzata da:
- adenoma paratiroideo;
- adenoma pituitario;
- tumori degli organi riproduttivi, come il cancro ai testicoli, il carcinoma cervicale neuroendocrino;
- tumori surrenali e renali.
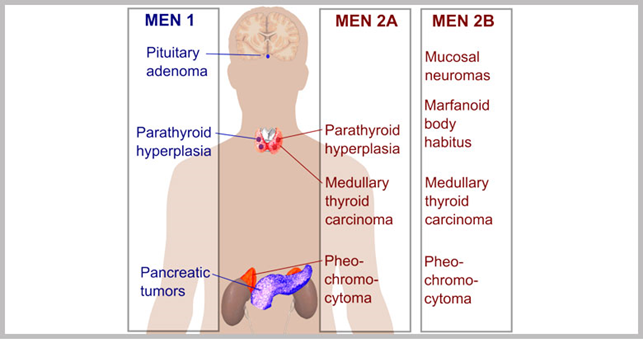
Immagine 1. Manifestazioni cliniche delle MEN
Tipi
- Di seguito vengono riportati i tipi di tumore e la penetranza tumorale stimata nella neoplasia endocrina multipla di tipo 1:
- adenoma paratiroideo nel 90% dei casi;
- adenoma ipofisario anteriore nel 30%-40%, la maggior parte dei quali sono prolattinomi, seguiti da somatotropinomi, corticotropinomi e tumori non funzionali;
- tumori enteropancreatici nel 30%-70%, compresi
- tumore non funzionante e secernente polipeptidi pancreatici nel 20%-50%;
- gastrinoma nel 40%;
- insulinoma nel 10%;
- glucagonoma in < 1%;
- tumore secernente peptide intestinale vasoattivo (VIPoma) in < 1%;
- tumori associati
- tumori surrenali:
- tumore corticale surrenale nel 40%, i sottotipi includono:
- non funzionale (il più comune);
- con produzione di cortisolo (sindrome di Cushing);
- con produzione di aldosterone (non comune);
- feocromocitoma in < 1%;
- tumore corticale surrenale nel 40%, i sottotipi includono:
- tumore neuroendocrino gastrico (NET) nel 10%;
- NET broncopolmonare nel 2%, i cui sottotipi includono:
- ben differenziato (carcinoide tipico e atipico);
- scarsamente differenziato (carcinoma neuroendocrino a piccole e grandi cellule);
- NET timico nel 2% (di solito non funzionale);
- tumori non endocrini tra cui vi sono:
- angiofibroma nel 22%-88%;
- collagenoma nello 0%-70%;
- lipoma nel 30%-33%;
- meningioma in < 10%.
- tumori surrenali:
- In uno studio monocentrico condotto in Cina, i tipi di adenomi ipofisari riscontrati nei 54 pazienti con MEN1 valutati da marzo 2003 a gennaio 2017 sono:
- adenomi ipofisari non funzionanti in 26 pazienti (48,1%);
- prolattinomi in 15 pazienti (27,8%);
- adenomi secernente l’ormone della crescita (somatotropinoma) in 5 (9,3%) pazienti;
- adenoma cosecernente in 5 pazienti (9,3%);
- adenoma secernente l’ormone adrenocorticotropina (ACTH) in 3 pazienti (5,6%).
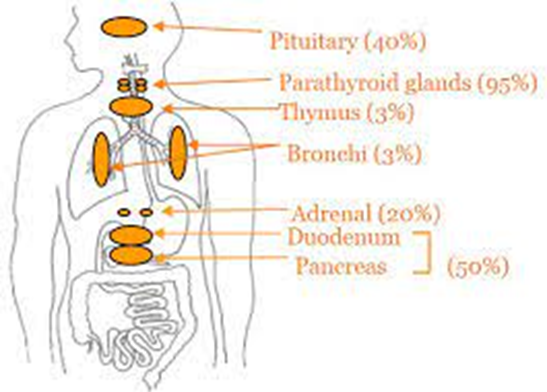
Immagine 2. Principali neoplasie della MEN1
Epidemiologia
Incidenza/Prevalenza
- La MEN 1 è una patologia rara, con una prevalenza stimata 1-10 casi su 100.000 persone.
Eziologia e patogenesi
Cause
- Si tratta di una malattia a trasmissione autosomica dominante causata da mutazioni germinali nel gene oncosoppressore MEN1 situato sul cromosoma 11q13.
- La forma sporadica o non familiare derivante da mutazioni de novo del gene MEN1 si verifica in circa il 10% dei pazienti.
Patogenesi
- Il gene della neoplasia endocrina multipla 1 (MEN1) codifica la proteina menina, una proteina nucleare coinvolta nella regolazione della trascrizione, della stabilità del genoma, della divisione cellulare e della proliferazione:
- se entrambi gli alleli MEN1 sono inattivati, MEN1 perde la sua attività di soppressore tumorale con conseguente sviluppo dei tumori associati a MEN 1;
- le varianti patogene ereditate o de novo della MEN1 in combinazione con una mutazione somatica acquisita (una mutazione puntiforme o più comunemente una delezione) provocano la perdita di eterozigosi e la crescita clonale.
Storia e fisica
Fisico
Fisico generale
- La maggior parte delle manifestazioni cliniche della MEN-1 sono legate alla produzione ormonale, quindi molti segni sono aspecifici, per esempio:
- Disidratazione;
- perdita di peso;
- ipertensione.
- È importante fare attenzione all’altezza nei bambini:
- l’aumento della velocità di crescita lineare può suggerire la diagnosi di somatotropinoma;
- vanno prese in considerazione anche altre diagnosi differenziali di Gigantismo.
Pelle
- Per quanto riguarda i possibili reperti cutanei a seconda del tipo di tumore, sono inclusi:
- eritema migratorio necrolitico nel caso del glucagonoma;
- acne, lividi facili, strie addominali (generalmente ampie e violacee) nel caso del corticotropinoma;
- sudorazione eccessiva, pelle arrossata in caso di un tireotropinoma;
- angiofibromi e collagenomi più frequenti nel caso di un gastrinoma.
HEENT
- I sintomi sono di solito legati alla secrezione ormonale da parte delle cellule tumorali o all’effetto massa determinato soprattutto in caso di tumori ipofisari (è infatti raro che l’iperparatiroidismo si presenti con un adenoma palpabile)
- qualsiasi tumore pituitario può causare difetti del campo visivo, oftalmoplegia;
- insulinoma si può manifestare con alterazione della vista (offuscamento) durante gli episodi di ipoglicemia;
- corticotropinoma si manifesta con “faccia lunare”;
- somatotropinoma determina segni di acromegalia come la protrusione della mascella, la sporgenza frontale e i lineamenti del viso più grossolani.
Cardiaco
- In caso di presenza di tachicardia o fibrillazione atriale in un paziente con MEN 1 si può ipotizzare la presenza di un tireotropinoma.
Addome
- L’obesità centrale può suggerire una sindrome di Cushing secondaria alla secrezione eccessiva di cortisolo da parte di un adenoma surrenale, un corticotropinoma o a causa della secrezione ectopica di ACTH da parte di un carcinoide bronchiale.
Schiena
- La “gobba di bufalo” o il cuscinetto di grasso dorsocervicale possono suggerire una sindrome di Cushing secondaria alla secrezione eccessiva di cortisolo da parte di un adenoma surrenale o di un corticotropinoma.
Neuro
- Possibili sintomia neurologici comprendono:
- letargia, psicosi più frequenti in caso di iperparatiroidismo;
- convulsioni, coma (a causa di ipoglicemia) nel caso di un insulinoma;
- iperattività, tremore nel caso di un tireotropinoma;
- psicosi, insonnia, debolezza muscolare prossimale nel caso di un corticotropinoma.
Diagnosi
Fare la diagnosi
- Sospettare la neoplasia endocrina multipla di tipo 1 (MEN1) in tutti i pazienti con ≥ 2 tumori endocrini associati alla MEN1.
- La diagnosi di neoplasia endocrina multipla di tipo 1 (MEN1) può essere basata su criteri clinici, familiari o genetici:
- criteri clinici – comparsa di ≥ 2 tumori endocrini primari associati alla MEN1:
- adenoma paratiroideo;
- adenoma pituitario anteriore;
- tumore enteropancreatico;
- criteri familiari – 1 tumore endocrino associato alla MEN1 e un parente di primo grado con MEN 1;
- criteri genetici – identificazione della mutazione germinale della MEN1, con o senza manifestazioni cliniche o biochimiche della MEN1.
- criteri clinici – comparsa di ≥ 2 tumori endocrini primari associati alla MEN1:
Panoramica dei test
- Il test genetico è raccomandato per identificare una mutazione germinale MEN1 in pazienti con sospetta MEN1 (pazienti con ≥ 2 tumori endocrini associati alla MEN1) (raccomandazione forte della Endocrine Society, evidenza di qualità moderata).
- Gli esami del sangue per individuare segni causati dai tumori associati alla MEN 1 includono:
- In caso di adenoma paratiroideo – aumento del calcio nel siero e ormone paratiroideo (PTH) inappropriatamente normale o aumentato;
- In caso di adenoma pituitario anteriore – aumento della prolattina, dell’ormone della crescita (GH), del fattore di crescita insulino-simile tipo 1 (IGF-1), dell’ormone adrenocorticotropo (ACTH) o del cortisolo;
- In caso di tumori neuroendocrini enteropancreatici (NET):
- Se gastrinoma – aumento della gastrina sierica a digiuno o test di provocazione della secretina positivo;
- Se insulinoma – ipoglicemia con contemporanea insulina elevata, C-peptide e proinsulina;
- Se VIPoma – VIP plasmatico aumentato a digiuno, e ipokaliemia, acloridria;
- Se altri tumori – aumento di glucagone, polipeptide pancreatico, ormone della crescita (GHRH), ormone della crescita (GH), somatostatina o cromogranina-A; bassa utilità di questi marcatori in diversi studi;
- In caso di tumori adrenocorticali – aumento del cortisolo o dell’aldosterone, diminuzione dell’ACTH o della renina, rispettivamente.
- L’imaging per localizzare i tumori può includere:
- risonanza magnetica (MRI) encefalo per l’identificazione dei tumori ipofisari;
- in caso di sospetti tumori enteropancreatici, si ricorre di solito a ecografia transaddominale e/o endoscopica, risonanza magnetica o tomografia computerizzata (TC); considerare anche la scintigrafia del recettore della somatostatina, l’iniezione selettiva di secretagoghi arteriosi (SASI) con campionamento venoso epatico;
- TC o RM torace se si sospetta un carcinoide bronchiale, carcinoide timico; TC o RM addome se tumori surrenali;
- Nel sospetto di tumori carcinoidi gastrici è utile l’esecuzione di un’endoscopia gastrica;
- In caso di tumori paratiroidei gli studi di imaging di solito non sono necessari, dal momento che nella MEN1, l’iperparatiroidismo è tipicamente trattato con paratiroidectomia a ghiandole multiple.
- Esami di campioni fecali possono aiutare a diagnosticare i NET enteropancreatici.
Gestione
- Non esiste tuttora uno studio randomizzato per il trattamento della neoplasia endocrina multipla di tipo 1 (MEN1); il trattamento varia in base al tipo di tumore presente.
- In caso di tumori paratiroidei:
- si raccomanda una paratiroidectomia totale (esplorazione bilaterale aperta con paratiroidectomia subtotale con rimozione di almeno 3,5 ghiandole); considerare una timectomia transcervicale concomitante;
- i trattamenti alternativi includono il cinacalcet e l’ablazione con etanolo.
- I tumori ipofisari anteriori vanno trattati in modo simile ai tumori ipofisari non associati alla MEN1:
- per il prolattinoma – gli agonisti della dopamina sono il trattamento di prima linea preferito;
- L’ipofisectomia chirurgica transfenoidale selettiva è il trattamento di prima linea preferito per altri tumori ipofisari e il trattamento di seconda linea per il prolattinoma farmaco resistente;
- La radioterapia è indicata in caso di tessuto tumorale residuo non resecabile.
- In caso di tumori neuroendocrini pancreatici (NET):
- considerare la chirurgia per i NET pancreatici non funzionanti (PNET) se > 1 cm o se la crescita è significativa nell’arco di 6-12 mesi;
- per la maggior parte dei pazienti con tumori neuroendocrini pancreatici funzionanti sintomatici tra cui glucagonoma, insulinoma e VIPoma:
- la chirurgia è raccomandata;
- se la chirurgia non è un’opzione, la gestione medica può includere analoghi della somatostatina, chemioterapia o embolizzazione dell’arteria epatica in caso di malattia metastatica;
- a differenza di altri PNET funzionanti sintomatici, la chirurgia è controversa per i gastrinomi associati a MEN-1 perché sono spesso multipli e si localizzano più frequentemente all’interno del duodeno:
- La sindrome di Zollinger-Ellison è spesso trattata con inibitori della pompa protonica come trattamento di prima linea nella maggior parte dei pazienti, con antagonisti del recettore H2 e analoghi della somatostatina come necessario;
- la chirurgia può essere curativa in casi selezionati con un centro chirurgico esperto, ed è tipicamente considerata quando il gastrinoma pancreatico è >2 cm di dimensione (in parte a causa del rischio di metastasi epatiche).
- In caso di altri tumori associati a MEN 1:
- tumori carcinoidi timici e bronchiali – la chirurgia è il trattamento di scelta;
- tumori carcinoidi gastrici – trattamento ottimale non chiaro, ma la resezione chirurgica è suggerita per tumori > 1 cm;
- tumori surrenali – chirurgia raccomandata per i tumori surrenali funzionanti e per i tumori non funzionanti con caratteristiche sospette di malignità per la gestione tipica dei tumori surrenali;
- si raccomanda uno screening regolare con esami del sangue annuali e studi di imaging regolari a partire dai 5 anni di età per individuare i tumori associati alla MEN 1 in caso di familiarità.
Complicazioni e prognosi
Complicazioni
- Complicazioni dell’iperparatiroidismo:
- nefrolitiasi;
- malattia renale cronica;
- osteoporosi e fratture.
- Complicazioni dei tumori ipofisari anteriori:
- infertilità, subfertilità e/o ipogonadismo (prolattinoma);
- acromegalia (somatotropinoma);
- Sindrome di Cushing (corticotropinoma);
- ipertiroidismo o tireotossicosi manifesta (tireotropinoma, raro);
- ipopituitarismo ed effetti di massa a seconda delle dimensioni.
- Complicazioni dei tumori neuroendocrini:
- malattia da ulcera peptica (gastrinoma o sindrome di Zollinger-Ellison);
- complicazioni neuroglicopeniche (insulinoma);
- diarrea acquosa, ipopotassiemia, sindrome di acloridria (WDHA) (VIPoma);
- eritema migratorio necrolitico, diabete mellito, perdita di peso (glucagonoma).
Prognosi
- La neoplasia endocrina multipla di tipo 1 (MEN1) è associata a un aumento del rischio di morte prematura e i tumori neuroendocrini timici o pancreatici sono associati a un maggior rischio di mortalità nei pazienti con MEN 1.
- Per la MEN 1 si raccomanda un imaging precoce e frequente e una sorveglianza con esami ematochimici.
- 2010 American Joint Committee on Cancer e 2006 European Neuroendocrine Tumor Society hanno stabilito dei sistemi di stadiazione del tumore che sembrano essere ugualmente efficaci per prevedere la sopravvivenza globale in pazienti sottoposti a tumori pancreatici resecabili non sindromici non funzionali.
- Rischio di malignità con i tumori legati alla MEN 1
- I tumori ipofisari associati alla MEN1 sono segnalati per diventare raramente maligni;
- i gastrinomi sono di solito multipli e maligni e metastatizzano rapidamente;
- L’80% dei glucagonomi associati a MEN-1 risulta essere maligno e metastatizzare al fegato;
- I VIPomi sono rari ma solitamente maligni e metastatizzano rapidamente, più frequentemente nel fegato;
- i tumori surrenali hanno un potenziale di malignità, maggiore in caso di tumori di grandi dimensioni;
- i tumori del timo hanno il più alto potenziale metastatico (42%-80%);
- i tumori bronchiali possono avere una buona prognosi.
Prevenzione e screening
Prevenzione
- non applicabile
MEN 2B
Definizione
- La neoplasia endocrina multipla di tipo 2B (MEN2B) è una sindrome tumorale autosomica dominante. È la forma più rara e aggressiva della MEN tipo 2 e rappresenta il 5%-10% dei casi.
- La MEN2B è causata da una mutazione germinale del gene RET situato sul cromosoma 10q11.2, più comunemente una mutazione a singolo nucleotide nel codone 918 dell’esone 16 del gene RET. L’incidenza della MEN2B è di circa 1 caso ogni 38.750.000 all’anno nella popolazione generale.
- Le caratteristiche cliniche classiche della MEN2B sono:
- carcinoma midollare della tiroide (si verifica nel 100% dei pazienti);
- feocromocitoma (si verifica in circa il 50% dei pazienti);
- facies tipica e anomalie oftalmologiche;
- ganglioneuromatosi del tratto gastrointestinale (si verifica in > 90% dei pazienti);
- malformazioni scheletriche come l’habitus marfanoide o la lordosi.
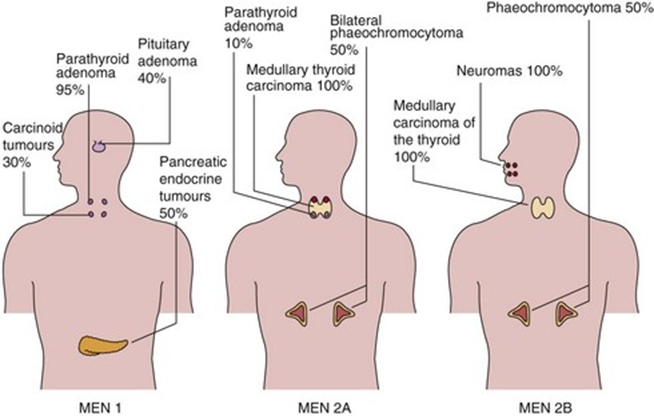
Immagine 01. Manifestazioni cliniche delle MEN.
Informazioni generali
Descrizione
- La neoplasia endocrina multipla di tipo 2B (MEN2B) è una sindrome tumorale autosomica dominante.
- La MEN2B è la forma più rara e aggressiva della neoplasia endocrina multipla di tipo 2 e rappresenta il 5%-10% dei casi di MEN.
- La MEN2B è caratterizzata da:
- carcinoma midollare della tiroide (si verifica nel 100% dei pazienti);
- feocromocitoma (si verifica in circa il 50% dei pazienti);
- facies atipica e anomalie oftalmologiche;
- ganglioneuromatosi del tratto gastrointestinale (si verifica in > 90% dei pazienti);
- malformazioni scheletriche come l’habitus marfanoide.
- Mentre la malattia paratiroidea non è presente nella MEN2B.
Chiamato anche
- MEN IIb
- MEN IIB
- MEN 2b
- sindrome del neuroma mucoso
- sindrome del neuroma mucoso multiplo
- Sindrome di Wagenmann-Froboese
Definizioni
- La neoplasia endocrina multipla di tipo 2 (MEN 2) comprende:
- MEN tipo 2A (sindrome di Sipple) (70%-95% dei pazienti con MEN 2), caratterizzata da:
- carcinoma midollare della tiroide (carcinoma midollare familiare riconosciuto come variante di MEN2A);
- feocromocitoma;
- iperparatiroidismo primario;
- associato a:
- lichen amiloidosico cutaneo (chiamato anche lichen planus amiloidosico);
- Malattia di Hirschsprung;
- MEN tipo 2B (circa il 5%-10% dei pazienti con MEN 2) caratterizzato da:
- carcinoma midollare della tiroide;
- feocromocitoma;
- associato a:
- ganglioneuromatosi;
- habitus marfanoide.
- MEN tipo 2A (sindrome di Sipple) (70%-95% dei pazienti con MEN 2), caratterizzata da:
- La neoplasia endocrina multipla di tipo 4 si caratterizza per la presenza di:
- adenoma paratiroideo;
- adenoma pituitario;
- tumori degli organi riproduttivi, come il cancro ai testicoli, il carcinoma cervicale neuroendocrino;
- tumori surrenali e renali.
Epidemiologia
- Risultano maggiormente colpiti i figli di un portatore con mutazione genetica (trattandosi di malattia autosomica dominante con il 50% di rischio di trasmissione).
Eziologia e patogenesi
Cause
- Mutazioni attivanti la linea germinale del proto-oncogene RET situato sul cromosoma 10q11:
- mutazione a singolo nucleotide nel codone 918 dell’esone 16 del gene RET (M918T) in > 95% dei casi;
- mutazione nel codone 883 (A883F) sull’esone 15 in < 5% dei casi;
- raramente i pazienti possono avere una doppia mutazione RET che appare in tandem sullo stesso allele che coinvolge il codone 804 e 806, 904, 805, o 781.
- Circa il 75% di tutti i casi sono sporadici con mutazioni de novo, tipicamente interessanti l’allele paterno.
Patogenesi
- L’oncogene RET codifica normalmente la produzione di un recettore tirosin-chinasi, che sembra trasdurre segnali per la crescita e la differenziazione in diversi tessuti in via di sviluppo, compresi quelli derivati dalla cresta neurale.
- Le mutazioni che causano la neoplasia endocrina multipla di tipo 2 (MEN tipo 2) portano ad un guadagno di funzione (attivazione) della tirosin-chinasi.
- Riguardo la correlazione genotipo-fenotipo per le mutazioni più comuni in MEN2B:
- i pazienti con mutazione A883F manifestano un carcinoma midollare della tiroide (MTC) meno aggressivo rispetto ai pazienti con mutazione M918T3;
- i pazienti con doppie mutazioni germinali RET sullo stesso allele (che coinvolgono il codone RET V804M e il codone RET Y806C, S904C, E805K o Q781R) sviluppano una MEN2B atipica all’età di 20-30 anni e tendono ad avere un MTC meno aggressivo rispetto ai pazienti con mutazione M918T3;
- i pazienti con MEN2B ereditaria sviluppano il MTC in età precoce rispetto a quelli che hanno mutazioni RET de novo e genitori fenotipicamente normali.

Tabella 1. Correlazione genotipo-fenotipo.
Storia e fisica
Presentazione clinica
- Quasi tutti i rapporti pubblicati sulla neoplasia endocrina multipla di tipo 2B (MEN tipo 2B) coinvolgono pazienti con mutazione RET nel codone 918 del gene; poco si sa sui pazienti con mutazione RET nel codone 883.
- Valutazione basata sullo stato noto di MEN tipo 2B:
- circa il 25% dei pazienti ha una MEN ereditaria di tipo 2B nota e può presentare un carcinoma midollare macroscopico della tiroide e metastasi linfonodali durante il primo anno di vita;
- circa il 75% ha mutazioni RET de novo e genitori fenotipicamente normali e di solito la diagnosi avviene in seguito al rilevamento di un nodulo tiroideo o per un fenotipo anormale (ganglioneuroma mucoso, habitus marfanoide, assenza di lacrime).
- La presentazione clinica nell’infanzia o nella prima infanzia può includere:
- presenza di neuromi della mucosa sulla superficie dorsale anteriore della lingua, del palato o della faringe;
- aspetto facciale caratteristico:
- le labbra diventano prominenti (o “gonfie”) nel tempo;
- noduli sottomucosi possono essere presenti sul bordo vermiglio;
- facies stretta e lunga;
- anomalie oftalmologiche:
- incapacità di produrre lacrime nell’infanzia;
- ispessimento ed estroflessione dei margini della palpebra superiore a causa di neuromi;
- ptosi lieve;
- nervi corneali spessi e prominenti possono essere visti all’esame con la lampada a fessura;
- malformazioni scheletriche:
- habitus corporeo marfanoide (si verifica in circa il 75% dei pazienti);
- pectum cavus;
- pectum excavatum;
- palato alto e arcuato;
- scoliosi;
- scivolamento dell’epifisi prossimanel del femore;
- ganglioneuromatosi diffusa del tratto gastrointestinale (riportata nel 93% in una serie di 28 pazienti):
- i sintomi includono distensione addominale, megacolon, costipazione intermittente o diarrea;
- alcuni pazienti richiedono un intervento chirurgico per l’ostruzione intestinale.
- Se non identificati dalla loro facies atipica e dall’habitus corporeo o dai sintomi gastrointestinali, i pazienti possono presentare sintomi o segni di:
- carcinoma midollare della tiroide:
- si sviluppa nel 100% dei pazienti;
- MTC macroscopico e metastasi linfonodali possono verificarsi durante il primo anno di vita nei pazienti con MEN2B ereditario;
- i primi sintomi possono includere:
- dolore al collo;
- massa palpabile del collo;
- linfonoadenopatia nel collo;
- diarrea come risultato dell’ipercalcitoninemia;
- di solito bilaterale e multicentrico;
- la diffusione metastatica è comune (fino al 70% ha metastasi linfonodali cervicali alla diagnosi);
- feocromocitoma:
- si sviluppa in circa il 50% dei pazienti;
- può svilupparsi già all’età di 8-12 anni;
- circa la metà sono multipli e spesso bilaterali;
- i segni e i sintomi includono mal di testa, palpitazioni, nervosismo, tachicardia e ipertensione.
- carcinoma midollare della tiroide:
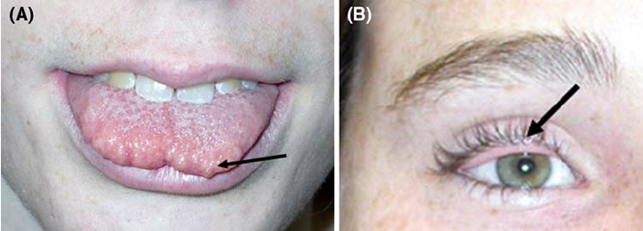
Immagine 02. Ccaratteristiche della facies tipica nei pazienti con MEN 2B.
Diagnosi
Fare la diagnosi
- Nella maggior parte dei pazienti la MEN2B viene diagnosticata quando il cancro midollare della tiroide è clinicamente evidente e troppo avanzato per essere curato.
- Lo screening è utile dopo la nascita di neonati a rischio o in pazienti che presentano caratteristiche cliniche, come:
- aspetto facciale caratteristico;
- anomalie oftalmologiche;
- malformazioni scheletriche;
- ganglioneuromatosi diffusa del tratto gastrointestinale;
- carcinoma midollare della tiroide (soprattutto ad insorgenza precoce);
- feocromocitoma.
- La diagnosi è confermata con un test genetico che confermi la presenza della mutazione del gene RET nelle cellule germinali.
- Il test genetico va eseguito nelle persone con:
- presunto MTC sporadico (ATA grado B);
- parenti di primo grado di pazienti con MTC ereditario provato (ATA Grado B);
- genitori di neonati o bambini con fenotipo classico di neoplasia endocrina multipla (MEN) di tipo 2B (ATA Grade B).
Panoramica dei test
- Nel paziente che presenta caratteristiche cliniche, l’obiettivo dei test è quello di diagnosticare il carcinoma midollare della tiroide e/o il feocromocitoma:
- nel sospetto di carcinoma midollare della tiroide eseguire:
- esami del sangue – misurazione concomitante di calcitonina e antigene carcinoembrionale (CEA) (ATA Grade B);
- biopsia e analisi immunoistochimica;
- in pazienti con MTC confermato citologicamente o istologicamente (ATA Grado C):
- valutare gli esami del sangue, compresi i livelli sierici di calcitonina e CEA e i test genetici per la ricerca della mutazione germinale del gene RET;
- escludere la presenza di feocromocitoma;
- eseguire studi di imaging preoperatorio;
- nel sospetto di un feocromocitoma eseguire:
- catecolamine e metaboliti delle catecolamine tramite raccolta di plasma o urine nelle 24 ore;
- imaging surrenale con tomografia computerizzata o risonanza magnetica (nel caso il risultato biochimico sia positivo (ATA Grado C).
- nel sospetto di carcinoma midollare della tiroide eseguire:
- Il test genetico per la mutazione del gene RET conferma la diagnosi e viene eseguito in un neonato a rischio subito dopo la nascita o in un paziente con chiaro fenotipo della neoplasia endocrina multipla di tipo 2B (MEN tipo 2B):
- Prima eseguire il test per la mutazione RET codone M918T (esone 16); se negativo, ricercare la mutazione del codone A883F (esone 15) (ATA Grado B). In realtà, il test per la ricerca delle mutazioni del gene RET può includere la ricerca di entrambi i codoni e quindi il test può avvenire allo stesso tempo;
- se non sono state identificate mutazioni, allora si procede con il sequenziamento dell’intera regione codificante di RET (ATA grado B).
- Valutazioni dopo la diagnosi iniziale di MEN2B se non già eseguite:
- fare riferimento all’endocrinologo e consultare un genetista clinico e/o un consulente genetico;
- test biochimici:
- calcitonina plasmatica;
- metanefrine plasmatiche;
- valutare la malattia metastatica nei pazienti con carcinoma midollare della tiroide eseguendo:
- tomografia computerizzata toracica e addominale con contrasto;
- risonanza magnetica del fegato o tomografia computerizzata (CT) dell’addome con protocollo epatico se la malattia nodale o la calcitonina > 400-500 pg/mL;
- scintigrafia ossea per escludere metastasi ossee.
Trattamento
Gestione del carcinoma midollare della tiroide
- Il trattamento standard per il carcinoma midollare della tiroide (MTC) è la tiroidectomia totale e la dissezione dei compartimenti linfonodali cervicali, a seconda dei livelli sierici di calcitonina e dei risultati ecografici.
- Per i pazienti con MTC e nessuna evidenza di metastasi ai linfonodi del collo o metastasi a distanza:
- eseguire una tiroidectomia totale (ATA grado B);
- eseguire la dissezione dei linfonodi nel compartimento centrale (livello VI) (grado B ATA);
- considerare la dissezione dei linfonodi nei compartimenti laterali (livelli II-V) in base ai livelli sierici di calcitonina (ATA Grado I).
- Per i pazienti con MTC confinato al collo e ai linfonodi cervicali (ATA Grado C):
- eseguire una tiroidectomia totale;
- eseguire la dissezione del compartimento linfonodale centrale (livello VI) e dei compartimenti laterali del collo coinvolti (livelli II-V);
- se l’imaging preoperatorio è positivo nel compartimento laterale ipsilaterale del collo, ma negativo nel compartimento controlaterale del collo, considerare la dissezione controlaterale del collo se il livello sierico basale di calcitonina > 200 pg/mL.
- Per i pazienti con malattia regionale o metastatica estesa, una chirurgia meno aggressiva nei compartimenti cervicali centrale e laterali può essere appropriata per preservare la parola, la deglutizione, la funzione paratiroidea e la mobilità della spalla; le opzioni per ottenere il controllo locale del tumore includono (ATA Grado C):
- radioterapia a fascio esterno (EBRT);
- terapia medica sistemica;
- altre terapie non chirurgiche.
- In caso di danno o devascolarizzazione durante la chirurgia delle ghiandole paratiroidi:
- durante una tiroidectomia totale per MTC, preservare le ghiandole paratiroidi normali in situ su un peduncolo vascolare (ATA Grado B);
- se tutte le ghiandole paratiroidi normali sono resecate o se nessuna appare vitale alla fine della procedura trapiantare frammenti di una ghiandola paratiroidea nel muscolo sternocleidomastoideo in pazienti con MEN2B (ATA Grado B).
- Dopo tiroidectomia unilaterale per presunto MTC sporadico, la tiroidectomia di completamento è raccomandata nei pazienti con una mutazione germinale RET, un elevato livello sierico di calcitonina postoperatorio, o studi di imaging che indicano MTC residuo (ATA Grado B).
- La gestione postoperatoria prevede:
- misurare l’ormone stimolante la tiroide (TSH) nel siero entro 4-6 settimane dopo l’intervento (ATA Grade B);
- somministrare una terapia sostitutiva dell’ormone tiroideo (levotiroxina) con l’obiettivo di mantenere il TSH sierico nel range eutiroideo (ATA Grade B);
- monitorare i livelli di calcio nel siero (ATA Grade B):
- somministrare calcio e vitamina D per via orale ai pazienti che sviluppano un’ipocalcemia sintomatica;
- la terapia sostitutiva cronica è indicata nei pazienti che non possono essere svezzati dai farmaci.
Gestione del feocromocitoma
- La resezione chirurgica del feocromocitoma dovrebbe precedere la chirurgia per il carcinoma midollare della tiroide (ATA Grado B).
- La surrenalectomia è indicata in pazienti con evidenza di feocromocitoma (ATA grado B):
- preferita la surrenalectomia laparoscopica o retroperitoneoscopica;
- considerare la surrenalectomia subtotale per preservare la funzione corticale surrenale come procedura alternativa.
- La surrenalectomia bilaterale non è consigliata a causa del rischio di insufficienza surrenalica e crisi addisoniana, che supera la preoccupazione per il rischio di sviluppare un feocromocitoma metacrono nella ghiandola surrenale controlaterale entro 10 anni.
- Un feocromocitoma diagnosticato durante la gravidanza dovrebbe essere resecato prima del terzo trimestre, se possibile (ATA Grado C).
Complicazioni e prognosi
Complicazioni
- Le complicazioni legate al carcinoma midollare della tiroide comprendono:
- ostruzione respiratoria;
- sanguinamento;
- metastasi ai linfonodi locali, fegato, polmone, osso.
- Le complicazioni legate all’aumento delle catecolamine (feocromocitoma) comprendono:
- Ipertensione;
- Ictus;
- infarto miocardico;
- insufficienza cardiaca.
- Le complicazioni legate alla ganglioneuromatosi del tratto gastrointestinale (presente in > 90% dei pazienti con neoplasia endocrina multipla di tipo 2B [MEN2B]) comprendono:
- Costipazione;
- Diarrea;
- Megacolon.
- Le complicazioni legate all’habitus marfanoide (presente nella maggior parte dei pazienti con MEN2B) comprendono:
- cifoscoliosi o lordosi;
- lassità articolare.
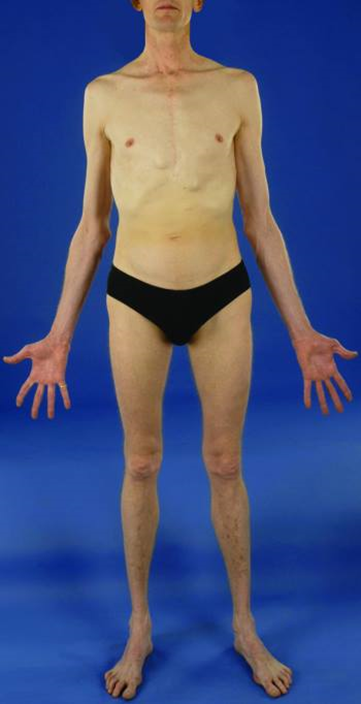
Immagine 03. Habitus marfanoide.
Prognosi
- Il carcinoma midollare della tiroide (MTC) associato alla neoplasia endocrina multipla di tipo 2B è clinicamente più aggressivo rispetto al MTC che si manifesta nella neoplasia endocrina multipla di tipo 2A:
- La storia naturale di MTC è variabile: da anni di malattia residua dormiente dopo l’intervento chirurgico alla malattia diffusa rapidamente progressiva e alla morte legata al tumore metastatico della tiroide;
- Le metastasi di MTC sono più comuni se i pazienti non sono stati sottoposti a una tiroidectomia precoce in giovane età;
- prima dell’intervento con tiroidectomia profilattica precoce, l’età media della morte era di 21 anni;
- I tassi di sopravvivenza a 10 anni per tutti i pazienti con MTC sono stati riportati al 61%-76%.
MEN2A
Panoramica e raccomandazioni
Definizione
- La neoplasia endocrina multipla di tipo 2A (MEN2A) è la più comune tra le 3 sindromi autosomiche dominanti caratterizzate dallo sviluppo di tumori endocrini; la sua incidenza è di circa 1 su 2 milioni all’anno nella popolazione generale.
- Ci sono 4 varianti di MEN2A, tra cui MEN2A classica, MEN2A con lichen amiloidosico cutaneo, MEN2A con malattia di Hirschsprung e MEN2A con cancro midollare familiare della tiroide.
- La MEN2A classica è la variante più comune e rappresenta il 70-95% dei casi di MEN2A. È caratterizzata dalla presenza di carcinoma midollare della tiroide associato alla presenza di un feocromocitoma (meno frequente, si verifica nel 40%-50% dei casi) e/o iperparatiroidismo (si verifica nel 20%-30% dei casi).
- La MEN2A è causata da mutazioni germinali del gene RET situato sul cromosoma 10q11.2.
Descrizione
- La neoplasia endocrina multipla di tipo 2A (MEN tipo 2A) è una sindrome tumorale autosomica dominante.
- Il tipo 2A rappresenta la maggior parte dei casi di MEN2 (dal 70% al 95%); il MEN2B rappresenta la restante percentuale dei casi.
- Esistono diverse varianti della MEN di tipo 2A; la MEN2A classica è la più comune ed è caratterizzata da:
- carcinoma midollare della tiroide (la prima manifestazione più comune);
- feocromocitoma;
- adenoma/iperplasia paratiroidea ( che si manifestano con iperparatiroidismo primario).
Chiamato anche
- MEN IIa
- MEN IIA
- MEN 2a
- MEN 2A
- MEN tipo 2
- MEN tipo II
- MEN2
- MEN 2
- adenomatosi endocrina multipla
- Sindrome di Sipple
Definizioni
- La neoplasia endocrina multipla di tipo 2 (MEN 2) comprende:
- MEN tipo 2A (sindrome di Sipple) (rappresenta il 70%-95% dei pazienti con MEN 2); si può manifestare con:
- carcinoma midollare della tiroide (carcinoma midollare familiare riconosciuto come variante di MEN2A);
- feocromocitoma;
- iperparatiroidismo primario;
- può associarsi a:
- lichen amiloidosico cutaneo (chiamato anche lichen planus amiloidosico);
- Malattia di Hirschsprung;
- MEN tipo 2B (comprende circa il 5%-10% dei pazienti con MEN 2); si manifesta con:
- carcinoma midollare della tiroide;
- feocromocitoma;
- può associarsi a:
- ganglioneuromatosi;
- habitus marfanoide.
- MEN tipo 2A (sindrome di Sipple) (rappresenta il 70%-95% dei pazienti con MEN 2); si può manifestare con:
- La neoplasia endocrina multipla di tipo 4 può manifestarsi con:
- adenoma paratiroideo;
- adenoma pituitario;
- tumori degli organi riproduttivi, come il cancro ai testicoli, il carcinoma cervicale neuroendocrino;
- tumori surrenali e renali.
Tipi
- Esistono quattro varianti di neoplasia endocrina multipla (MEN) di tipo 2A:
- MEN2A classico:
- È la variante più comune;
- È caratterizzato dalla presenza di carcinoma midollare della tiroide con meno frequentemente la presenza di feocromocitoma e/o iperparatiroidismo;
- MEN2A con lichen amiloidosico cutaneo;
- MEN2A con malattia di Hirschsprung;
- tumore midollare familiare della tiroide (famiglie o individui con mutazioni germinali RET che hanno un tumore midollare della tiroide ma né feocromocitoma né iperparatiroidismo).
- MEN2A classico:
Epidemiologia
Chi è più colpito
- Colpisce principalmente la prole di un portatore con mutazione genetica (malattia autosomica dominante con il 50% di rischio di trasmissione).
Eziologia e patogenesi
Cause
- La MEN2A è causata da mutazioni germinali attivanti del proto-oncogene RET situato sul cromosoma 10q11.2; le mutazioni più comuni includono:
- residui di cisteina nel codone 634 dell’esone 11;
- residui di cisteina nei codoni 609, 611, 618 e 620 sull’esone 10.
Patogenesi
- L’oncogene RET codifica normalmente la produzione di un recettore tirosin-chinasi, che sembra trasdurre segnali per la crescita e la differenziazione in diversi tessuti in via di sviluppo, compresi quelli derivati dalla cresta neurale.
- Le mutazioni che causano la neoplasia endocrina multipla di tipo 2 (MEN 2) portano ad un guadagno di funzione (attivazione) della tirosin-chinasi.
- MEN2A associato con lichen amiloidosico cutaneo è associato a notalgia paresthetica, una neuropatia sensoriale che coinvolge i nervi spinali dorsali.
- Le correlazioni genotipo-fenotipo sono stabilite e utilizzate per la classificazione del rischio cioè per determinare i tempi e l’estensione della tiroidectomia.
Storia e fisica
Presentazione clinica
- La MEN 2A può essere asintomatica, soprattutto quando diagnosticata in seguito a esecuzione dello screening per i parenti di primo grado di pazienti con neoplasia endocrina multipla di tipo 2A (MEN2A).
- I pazienti con MEN2A classica possono presentare:
- carcinoma midollare della tiroide:
- si verifica in oltre il 90% dei casi e di solito è la prima manifestazione della MEN2A;
- in genere si presenta con una massa o un dolore al collo prima dei 35 anni, anche se può presentarsi in età più avanzata in pazienti con carcinoma midollare della tiroide familiare;
- i primi sintomi possono includere:
- dolore al collo;
- massa palpabile del collo;
- linfonodi gonfi nel collo;
- diarrea come risultato dell’ipercalcitoninemia;
- di solito bilaterale e multicentrico;
- la diffusione metastatica è comune (fino nel 70% dei casi sono presenti metastasi ai linfonodali cervicali alla diagnosi);
- feocromocitoma:
- si verifica in circa il 40%-50% dei casi ma riportato alla presentazione iniziale nel 13%-27%;
- si presenta tipicamente nella terza o quarta decade di vita e la diagnosi è solitamente fatta in concomitanza o in seguito alla diagnosi di cancro midollare della tiroide;
- i primi sintomi possono includere:
- mal di testa;
- palpitazioni;
- nervosismo;
- sudorazione;
- ipertensione;
- di solito multicentrico, bilaterale e confinato alla ghiandola surrenale;
- tipicamente è associato ad una diffusa iperplasia nodulare della midollare del surrene; la trasformazione maligna si verifica in < 1% dei casi;
- metastasi rare;
- i pazienti che alla diagnosi presentano un feocromocitoma unilaterale di solito sviluppano un feocromocitoma controlaterale entro 10 anni;
- rispetto ai feocromocitomi isolati, i feocromocitomi della MEN2A vengono diagnosticati in un’età più precoce, presentano una sintomatologia meno evidente ed è più probabile che siano bilaterali;
- iperparatiroidismo:
- raramente è il sintomo d’esordio di una MEN2A; di solito si presenta dopo la diagnosi di carcinoma midollare della tiroide;
- circa il 20%-30% dei pazienti con MEN2A sviluppa l’iperparatiroidismo ad un’età media di insorgenza di 38 anni;
- la maggior parte degli individui con iperparatiroidismo sono asintomatici;
- può manifestarsi con ipercalciuria e calcoli renali;
- le ghiandole paratiroidi possono essere ingrandite; la patologia varia da un singolo adenoma a una marcata iperplasia;
- carcinoma midollare della tiroide:
- pazienti con MEN2A e lichen amiloidosico cutaneo:
- può anche presentarsi con:
- lesioni cutanee, soprattutto nella regione scapolare della schiena corrispondente ai dermatomi T2-T6;
- prurito intenso che migliora con l’esposizione al sole e peggiora durante i periodi di stress (sintomo classico);
- lesioni iperpigmentate, che possono svilupparsi successivamente, probabilmente a causa di graffi;
- il lichen amiloidosico cutaneo può presentarsi prima dell’insorgenza di un cancro midollare alla tiroide clinicamente evidente;
- può anche presentarsi con:
- pazienti con MEN2A e malattia di Hirschsprung:
- in genere si presentano con la malattia di Hirschsprung poco dopo la nascita;
- può presentarsi in età adulta con sintomi del colon suggestivi della malattia di Hirschsprung.
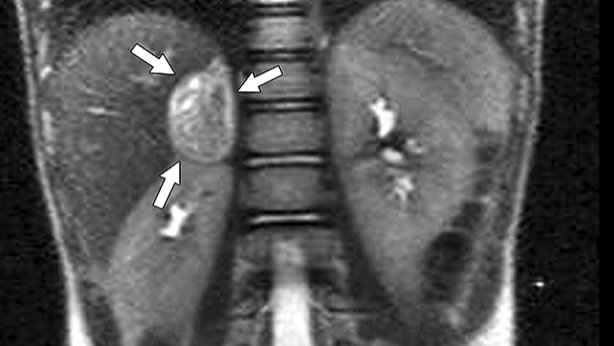
Immagine 02. Riscontro RMN di feocromocitoma.
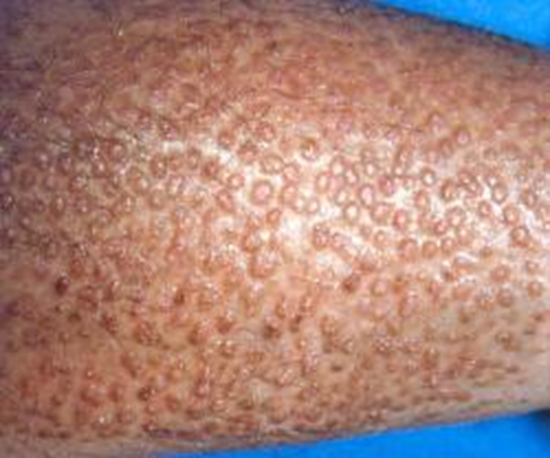
Immagine 03. Lichen amiloidosico cutaneo.
Esame obiettivo
Esame obiettivo generale
- La maggior parte dei pazienti presenta un esame obiettivo normale.
- I pazienti con feocromocitoma possono presentare:
- pressione sanguigna elevata;
- tachicardia.
Pelle
- Possono essere presenti lesioni iperpigmentate (soprattutto nei pazienti con MEN2A e lichen amiloidosico cutaneo).
Collo
- I pazienti con carcinoma midollare della tiroide possono avere:
- massa tiroidea palpabile;
- linfoadenopatia cervicale.
Diagnosi
Fare la diagnosi
- È corretto sospettare una neoplasia endocrina multipla di tipo 2A (MEN2A) se sono presenti (nel paziente o nei parenti di primo grado) almeno una o più delle 3 caratteristiche cliniche classiche:
- carcinoma midollare della tiroide (MTC);
- feocromocitoma;
- iperparatiroidismo.
- La diagnosi è clinicamente stabilita se sono presenti ≥ 2 sintomi caratteristici tra MTC, feocromocitoma o adenoma/iperplasia paratiroidea in un singolo individuo o in parenti stretti.
- Considerare l’esecuzione di test genetici in persone con:
- presunto MTC sporadico (ATA grado B);
- parenti di primo grado con MTC ereditario provato (ATA grado B);
- una diagnosi di lichen amiloidosico cutaneo (ATA grado B);
- una diagnosi di iperplasia primaria delle cellule C o MTC;
- una diagnosi clinica di MEN 2, che abbiano o meno una storia familiare positiva.
Panoramica dei test
- Nel paziente che presenta una qualsiasi delle caratteristiche cliniche classiche, l’obiettivo dei test è quello di diagnosticare il carcinoma midollare della tiroide, il feocromocitoma o l’iperparatiroidismo primario:
- nel sospetto di un carcinoma midollare della tiroide (MTC) andranno eseguiti:
- esami del sangue comprendenti misurazione simultanea di calcitonina e antigene carcinoembrionale (CEA) (ATA Grade B);
- biopsia e analisi immunoistochimica;
- in pazienti con MTC confermato citologicamente o istologicamente (ATA Grado C):
- valutare gli esami del sangue, compresi i livelli sierici di calcitonina e CEA e i test genetici per una mutazione germinale RET;
- escludere la presenza di feocromocitoma e iperparatiroidismo;
- eseguire studi di imaging preoperatorio;
- nel sospetto di un feocromocitoma è utile eseguire:
- il dosaggio dei metaboliti delle catecolamine (metanefrine) nel plasma o nella raccolta di urine di 24 ore;
- un’indagine di imaging surrenale con tomografia computerizzata o risonanza magnetica, indicata soprattutto se il risultato biochimico è positivo (ATA Grado C);
- nel sospetto di iperparatiroidismo, questo va confermato mediante il dosaggio sierico di calcio e ormone paratiroideo.
- nel sospetto di un carcinoma midollare della tiroide (MTC) andranno eseguiti:
- Il test genetico per la mutazione germinale del gene RET conferma la diagnosi e viene eseguito nel paziente con sospetta diagnosi clinica o come parte dello screening nei parenti di primo grado; prevede:
- Un test iniziale con analisi singola o multilivello per individuare mutazioni RET nell’esone 10 (codoni 609, 611, 618 e 620), nell’esone 11 (codoni 630 e 634) e negli esoni 8, 13, 14, 15 e 16 (ATA Grado B);
- L’analisi dei codoni per identificare il sottotipo di neoplasia endocrina multipla (MEN) e il rischio di cancro alla tiroide.
- In seguito alla diagnosi iniziale di MEN2A:
- fare riferimento all’endocrinologo e consultare un genetista clinico e/o un consulente genetico;
- eseguire test biochimici quali:
- calcitonina plasmatica;
- catecolamine e metanefrine plasmatiche;
- calcio sierico e ormone paratiroideo;
- valutare la malattia metastatica nei pazienti con carcinoma midollare della tiroide, mediante l’impiego di:
- tomografia computerizzata toracica e addominale con contrasto;
- risonanza magnetica del fegato o TAC dell’addome con protocollo epatico se la malattia nodale o la calcitonina > 400 pg/mL.
- Per i pazienti con diagnosi citologica o istologica di MTC, eseguire esami del sangue compresi i livelli sierici di calcitonina e CEA e test genetici per l’identificazione di mutazioni germinali del gene RET (ATA Grado C).
Trattamento
Gestione del carcinoma midollare della tiroide
- Il trattamento standard per il carcinoma midollare della tiroide (MTC) è la tiroidectomia totale e la dissezione dei compartimenti linfonodali cervicali, a seconda dei livelli sierici di calcitonina e dei risultati ecografici.
- Per i pazienti con MTC e nessuna evidenza di metastasi ai linfonodi del collo o metastasi a distanza:
- eseguire una tiroidectomia totale (ATA grado B);
- eseguire la dissezione dei linfonodi nel compartimento centrale (livello VI) (grado B ATA);
- considerare la dissezione dei linfonodi nei compartimenti laterali (livelli II-V) in base ai livelli sierici di calcitonina (ATA Grado I).
- Per i pazienti con MTC confinato al collo e ai linfonodi cervicali (ATA Grado C):
- eseguire una tiroidectomia totale;
- eseguire la dissezione del compartimento linfonodale centrale (livello VI) e dei compartimenti laterali del collo coinvolti (livelli II-V);
- se l’imaging preoperatorio è positivo nel compartimento laterale ipsilaterale del collo, ma negativo nel compartimento controlaterale, considerare la dissezione controlaterale del collo se il livello sierico basale di calcitonina > 200 pg/mL.
- Per i pazienti con malattia regionale o metastatica estesa, una chirurgia meno aggressiva nel collo centrale e laterale può essere appropriata per preservare la parola, la deglutizione, la funzione paratiroidea e la mobilità della spalla; le opzioni per ottenere il controllo locale del tumore includono (ATA Grado C):
- radioterapia a fascio esterno (EBRT);
- terapia medica sistemica (cabozantinib, vandetanib o terapie sperimentali);
- altre terapie non chirurgiche.
- Per quanto riguarda la gestione delle ghiandole paratiroidi normali resecate o devascolarizzate durante la chirurgia tiroidea:
- durante una tiroidectomia totale per MTC, cercare di preservare le ghiandole paratiroidi normali in situ su un peduncolo vascolare (ATA Grado B);
- se tutte le ghiandole paratiroidi normali vengono resecate o se nessuna appare vitale alla fine della procedura (ATA Grado B) vi sono due possibili soluzioni:
- trapianto di schegge di una ghiandola paratiroidea nel muscolo sternocleidomastoideo in pazienti con MEN2A e una mutazione RET raramente associato con iperparatiroidismo;
- trapiantare schegge di una ghiandola paratiroidea in un letto muscolare eterotopico in pazienti con MEN2A e una mutazione RET associata ad un’alta incidenza di iperparatiroidismo.
- Dopo tiroidectomia unilaterale per presunto MTC sporadico, la tiroidectomia di completamento è raccomandata nei pazienti con una mutazione germinale RET, un elevato livello di calcitonina sierica postoperatoria o studi di imaging che indicano MTC residuo (ATA Grado B).
- Nella gestione postoperatoria è importante:
- misurare l’ormone stimolante la tiroide (TSH) nel siero entro 4-6 settimane dopo l’intervento (ATA Grade B);
- somministrare una terapia sostitutiva dell’ormone tiroideo (levotiroxina) con l’obiettivo di mantenere il TSH sierico nel range eutiroideo (ATA Grade B);
- monitorare i livelli di calcio nel siero (ATA Grade B)
- somministrare calcio e vitamina D per via orale ai pazienti che sviluppano un’ipocalcemia sintomatica;
- la terapia sostitutiva cronica è indicata nei pazienti che non possono essere svezzati dai farmaci.
Gestione del feocromocitoma
- La resezione chirurgica del feocromocitoma dovrebbe precedere l’intervento per il carcinoma midollare della tiroide o il trattamento dell’iperparatiroidismo primario (ATA Grade B).
- La surrenalectomia è indicata in pazienti con evidenza di feocromocitoma (ATA grado B):
- È preferita la surrenalectomia laparoscopica o retroperitoneoscopica;
- considerare la surrenalectomia subtotale per preservare la funzione corticale surrenale come procedura alternativa.
- La surrenalectomia bilaterale non è consigliata a causa del rischio di insufficienza surrenalica e crisi addisoniana, che supera la preoccupazione per il rischio di sviluppare un feocromocitoma metacrono nella ghiandola surrenale controlaterale entro 10 anni.
- Un feocromocitoma diagnosticato durante la gravidanza dovrebbe essere resecato prima del terzo trimestre, se possibile (ATA Grado C).
Gestione dell’iperparatiroidismo
- Se la diagnosi di iperparatiroidismo è stabilita prima o al momento della tiroidectomia:
- Trattare l’iperparatiroidismo chirurgicamente al momento della tiroidectomia;
- considerare la resezione solo delle ghiandole paratiroidi visibilmente ingrandite (ATA Grado C), con monitoraggio intraoperatorio dell’ormone paratiroideo per documentare la rimozione completa del tessuto paratiroideo iperfunzionante;
- se tutte e 4 le ghiandole sono visibilmente ingrandite, le opzioni includono (ATA Grado C):
- paratiroidectomia subtotale, lasciando un pezzo di 1 ghiandola in situ su un peduncolo vascolare;
- paratiroidectomia totale con autotrapianto eterotopico.
- Se l’iperparatiroidismo si sviluppa dopo una precedente tiroidectomia considerare di (ATA Grado C):
- eseguire studi di localizzazione preoperatori prima di ripetere la chirurgia del collo;
- resecare solo le ghiandole paratiroidi ingrossate, lasciando in situ le paratiroidi di dimensioni normali
- se c’è evidenza di una precedente resezione di 3 ghiandole paratiroidi, lasciare una porzione di ghiandola allargata in situ con un adeguato apporto di sangue o un innesto in un sito eterotopico.
Complicazioni e prognosi
Complicazioni
- Complicazioni legate al carcinoma midollare della tiroide:
- ostruzione respiratoria;
- sanguinamento;
- metastasi ai linfonodi locali, fegato, polmone, osso.
- Complicazioni legate all’aumento delle catecolamine (feocromocitoma):
- Ipertensione;
- Ictus;
- infarto miocardico (MI);
- insufficienza cardiaca.
- Complicazioni legate all’ipercalcemia secondaria (iperparatiroidismo):
- Nefrolitiasi;
- osteoporosi o fratture.
Prognosi
- Carcinoma midollare della tiroide (MTC):
- la storia naturale del MTC è variabile e va da anni di malattia residua dormiente dopo l’intervento chirurgico a una malattia disseminata rapidamente progressiva;
- una recidiva è riportata in circa il 50% dei pazienti con MTC sottoposti a tiroidectomia totale e dissezioni linfonodali del collo;
- Sopravvivenza a 10 anni per i pazienti con MTC 61%-76%.
- I pazienti con neoplasia endocrina multipla di tipo 2A (MEN2A) hanno un tasso di sopravvivenza migliore rispetto ai pazienti con tumori simili ma sporadici.
- L’analisi precoce della mutazione del gene RET e la tiroidectomia profilattica prima della comparsa dei sintomi migliorano la qualità della vita e prolungano l’aspettativa di vita.